Hiper- ve Hipolipoproteinemiler
Hiperlipidemi
• Başlıca plazma lipidleri olan kolesterol ve
trigliseridlerden biri ya da her ikisinin arttığı durum
• Serum lipoprotein konsantrasyonlarının artışına
hiperlipoproteinemi denir
Fredrickson Sınıflandırması (1965)
• Hiperlipidemileri 5 sınıfa ayırır
• ApoB içeren lipoproteinlerin, açlık durumunda, kağıt
elektroforezindeki görünümlerini esas alır
• Her fenotip belli bir hastalığı değil, aynı
lipoproteinleri etkileyerek aynı lipid paternine sebep
olan değişik bozuklukları temsil etmektedir
• Tedavi aynı fenotipe giren bütün bozukluklar için
aynıdır
• HDL’yi dikkate almamaktadır
WHO Sınıflandırması
Tip 1
Tip 2a
Tip 2b
Tip 3
Tip 4
Tip 5
Hiperlipoproteinemi (Hiperşilomikronemi)
″
″
″
″
″
(Hiperbetalipoproteinemi)
(Kombine Hiperlipoproteinemi)
(Disbetalipoproteinemi)
(Hiperprebetalipoproteinemi)
(Mixed Hiperlipoproteinemi)
• WHO sınıflandırması da hiperlipoproteineminin genetik temeli
mekanizması, aile taramasına gerek olup olmadığı, tedavi
ilkeleri ve
• Hipolipoproteinemiler ve HDL anomalileri konusunda bilgi
vermez
• Günümüzde bu bozuklukları sınıflandıracak ideal bir sistem
bulunmamaktadır
• Lipoprotein metabolizmasındaki enzimleri, reseptörleri veya
apolipoproteinleri kodlayan genlerdeki mutasyonların yanısıra
yaşam biçimi veya sekonder sebepler de söz konusu olabilir
• İzleyen sınıflandırma laboratuvar bulgularına göre yapılmıştır
Yüksek Total Kolesterol + Yüksek LDL-kolesterol
• Polijenik (Nonfamilyal) Hiperkolesterolemi
• Familyal Hiperkolesterolemi (FH)
• Familyal Defektif Apo B-100 (FDB)
• Sitosterolemi
• Otozomal Dominant Hiperkolesterolemi
• Otozomal Resessif Hiperkolesterolemi (ARH)
Polijenik (Nonfamilyal) Hiperkolesterolemi
• Hiperkolesterolemi Batı toplumlarında daha sıktır,
toplumdaki hiperkolesteroleminin %85’i bu gruba girer
• Monojenik ve sekonder bozuklukların dışındaki primer
bozukluklar bu gruba girer
• Sebep multifaktöryeldir
• Çevresel faktörler, diyet, diyetteki doymuş yağ
miktarı önemlidir
Familyal Hiperkolesterolemi (FH)
• Etkilenip de tedavi edilmeyen kişilerdeki ateroskleroz
gelişme eğilimi nedeni ile hiperlipidemiler içinde en ciddi
olanıdır. Sebep LDLR genindeki mutasyonlardır
• Heterozigotlarda belirgin hiperkolesterolemi (yüksek LDLkolesterol) ve prematür kardiyovasküler hastalık riski
vardır
• Heterozigot FH en sık rastlanan kalıtsal hastalıklardan
biridir (1/500)
• Tedavi edilmemiş heterozigotların %70’inde parmakların
ekstansör tendonlarında ve Achille tendonunda görülen
ksantomlar klinik tanıya yardım eder
LDL Reseptörü (LDLR)
• Transmembran bir proteindir
• Hem apo B-100 hem de apo E’yi bağlar
• LDL apo B-100, IDL ise apo E aracılığı ile bağlanır
• LDLR’nin 5 bölgesi vardır
• Ligand bağlayıcı bölge amino terminalindedir
• Karboksi terminali sitoplazmik bölgeyi oluşturur,
reseptörü coated pit’e yönlendirir
• LDLR’nin ekspresyonu feedback kontrol altındadır
• Denge durumunda reseptör sayısı, hücre büyümesi ve
kayıpları dengelemek için gerekli kolesterolü sağlayacak
kadardır
• Bu kontrol membrana bağlı transkripsiyon faktörleri
(SREBP), Insig ve SCAP ile sağlanır
LDL Reseptör Geninde Mutasyon
• Sınıf 1 mutasyonlar: En sık rastlanan; LDL reseptör
proteini ER’da sentezlenemez
• Sınıf 2 mutasyonlar: ER’da sentez normaldir, Golgi’ye
taşınamaz
• Sınıf 3 mutasyonlar: Reseptör hücre yüzeyine taşınır ama
LDL’yi bağlayamaz
• Sınıf 4 mutasyonlar: Reseptör-ligand kompleksi “coated pit”
te yerleşemez
• Sınıf 5 mutasyonlar: Reseptörler hücre membranına geri
dönemez
Familyal Defektif Apo B-100 (FDB)
• Apo B-100’ün, LDLR bağlayıcı bölgesini etkileyen gen
mutasyonu
• LDL’nin, LDL reseptörüne affinitesi azalır
• Fenotip olarak FH’ye çok benzer
• Plazma total ve LDL-kolesterol düzeyleri artmıştır
• FH’ye göre artış daha az, tendon ksantomları daha
nadirdir
Sitosterolemi
• Çok ender görülür
• Fitosteroller (bitki sterolleri) emilerek plazma ve
periferik dokularda birikir
• Sebebi ABCG8 veya ABCG5 genlerindeki mutasyonlardır
• Bozukluk nedeniyle bitki sterolleri uygun kanal oluşturulup
bağırsak lumenine pompalanamaz, karaciğer emilen
sterolleri safraya sekrete edemez
• Ksantomlar, prematür ateroskleroz, angina, MI, ani ölüm
görülebilir
• Hemolitik anemi ilk bulgu olabilir
• Sterolden fakir diyet ve ezetimib (sterol emilim
inhibitörü) kullanılır
Otozomal Dominant Hiperkolesterolemi
(PCSK9’da Fonksiyonu Artıran Mutasyonlar)
• Fenotip olarak klasik FH ve FDB’den ayırt edilemeyen
ailelerin incelenmesi ile bulunmuştur
• Proprotein konvertaz subtilisin keksin 9 (PCSK9) genindeki
mutasyonlar sonucudur
• PCSK9’un LDL reseptörlerini yıkma hızını arttıran bu
mutasyonlar hücre yüzeyinde daha az reseptör
ekspresyonuna ve sonuçta plazmadan hücre içine LDL
alımının azalmasına sebep olur
• Plazma LDL-kolesterol düzeyleri ve damar hastalığı klasik FH
hastalarından daha ciddidir
Otozomal Resessif Hiperkolesterolemi (ARH)
• LDLRAP1 geninde mutasyon sonucudur, LDLR-LDL
kompleksinin internalizasyonu bozulmuştur
•
LDLRAP1 geninin protein ürünü adaptör proteindir
• LDLRAP1 LDL’nin reseptörüne bağlandıktan sonraki
endositozunu sağlar
• Fenotip olarak homozigot FH’lilere benzerler
• FH’deki gibi koroner ve karotis arterlerinde prematür
ateroskleroz gelişir
Yüksek Trigliserid + Normal Kolesterol
• Diyabetik Dislipidemi
• Familyal Hipertrigliseridemi
• Lipoprotein Lipaz Eksikliği (Şilomikronemi
Sendromu)
• Apolipoprotein C-II Eksikliği
• Apolipoprotein C-III Fazlalığı
• Apolipoprotein A-V Eksikliği
Diyabetik Dislipidemi
• Tip 2 diyabet hastalarında görülen aterojenik
dislipidemidir (yüksek trigliserid, düşük HDL, küçükyoğun LDL)
• Tedavide primer hedef LDL-kolesteroldür
Familyal Hipertrigliseridemi
• Hastaların çoğunda orta derecede, daha azında ise ciddi
bir trigliserid (VLDL) artışı vardır
• VLDL partikülleri normalden daha büyüktür, karaciğerde
trigliserid sentezinin artmış olduğunu düşündürür
• Mekanizma bilinmemekle birlikte çevresel faktörlerin
genetik varyantlarla etkileşiminin etkili olduğu
düşünülmektedir
• Şilomikronemi sendromu dışındaki sebeplere bağlı olanlar
erişkin yaşlarda ortaya çıkar
Şilomikronemi Sendromu
• Açlık durumunda plazmada şilomikronlar vardır
• Plazma trigliserid düzeyi 1000 mg/dL
• HDL-kolesterol düzeyi çok düşüktür
• Tekrarlayan karın ağrısı atakları (akut pankreatit)
• Hepatosplenomegali, erüptif ksantomlar, lipemia retinalis
Erüptif Ksantoma
Lipemia retinalis
Lipoprotein Lipaz Eksikliği
(Tip 1 Hiperlipoproteinemi, Hiperşilomikronemi)
• Ender görülen (1 milyonda 1) otozomal resessif bozukluk
• Genellikle çocukluk döneminde belirti verir
• Postheparin LPL aktivite tayini ile tanı konur
• Enzim konsantrasyonu yüksek olsa bile katalitik
aktivitesi yoktur
• Yüksek plazma şilomikron ve buna bağlı trigliserid
düzeyleri (açlıkta >1000 mg/dL, yemek sonrası > 10 000
mg/dL)
Apolipoprotein C-II Eksikliği
• Klinik semptomlar LPL eksikliğindeki gibidir ama daha
hafiftir ve daha geç ortaya çıkar
• LPL aktivatörü apo C-II, vakaların %50’sinde mevcuttur
ama LPL’ı aktive etmez
• Postheparin LPL aktivitesi düşüktür ama apo C-II
eklenince artar
• Trigliserid düzeyleri yükselir (500-10 000 mg/dL)
• Pankreatit atakları, tekrarlayıcı karın ağrıları
• Plazma transfüzyonu ile apo C-II sağlanabilir
Apolipoprotein C-III Fazlalığı
• Hem LPL aktivitesini bozar, hem de apo B’nin karboksi
terminaline bağlanarak LDLR’ye bağlanmasını engeller
• Apo C-III düzeyleri tip 2 DM, hiperbilirubinemi,
böbrek yetersizliği ve hipotiroidide yükselir
• Artan yaş, alkol tüketimi, oral kontraseptif kullanımı
da düzeyleri artırır
• Genetik faktörler de söz konusudur
Apolipoprotein A-V Eksikliği
• VLDL’nin organize olmasında rol oynadığı düşünülmektedir
• LPL’ye bağlı trigliserid hidrolizini aktive ettiği de
düşünülmektedir
• Düşük düzeyleri hipertrigliseridemiye sebep olur
Yüksek Kolesterol + Yüksek Trigliserid
• Familyal Kombine Hiperlipidemi (Tip 2B)
• Edinilmiş Kombine Hiperlipidemi
• Disbetalipoproteinemi (Remnant Hiperlipoproteinemi)
(Tip 3)
• Hepatik Lipaz Eksikliği
• Kolesterol-7-alfa-hidroksilaz Eksikliği
Familyal Kombine Hiperlipidemi
• İskemik kalp hastalığı ile başvuran kişilerde en sık
rastlanan lipid bozukluğu (1:100)
• Erişkin yaşa kadar genellikle belirti vermez
• Total kolesterol (apo B-100 ve LDL) ve/veya trigliserid
(VLDL) düzeyleri artmıştır
• Apo B-100 fazla miktarda yapılmaktadır
• Ailenin farklı bireylerinde farklı fenotipler gözlenir,
aynı kişide patern değişebilir
• **LDL küçük ve yoğun, HDL-kolesterol düşük,
trigliserid yüksektir (aterojenik lipid profili)
• Hastalar genellikle obezdir, karaciğerde müsait
trigliserid artmıştır
Edinilmiş Kombine Hiperlipidemi
• Metabolik sendromlu kişilerde sık görülür
• Tip 2 DM, hipertansiyon, santral obezite, KKH’nı
içeren bir sendromdan kaynaklanmaktadır
• Plazmadaki yüksek serbest yağ asidi düzeyi
karaciğerde fazla miktarda VLDL sentezine sebep olur
• VLDL, LDL’ye olgunlaşır
• LDL ve VLDL artar
Remnant Hiperlipoproteinemi (Tip 3)
• Disbetalipoproteinemi de denir
• VLDL fraksiyonu kolesterolden zengindir
• Sebep, karaciğer ve bağırsak kaynaklı kalıntı
partiküllerindeki artıştır
• Elektroforezde birbirinden ayrı pre-β ve β bantları
yerine geniş bir β bantı görülür
• Hem kolesterol hem de trigliserid düzeyleri artmıştır
• Hastalar apo E-2 için homozigottur
• Fenotipin ortaya çıkması için obezite, diyabet, aşırı alkol
tüketimi, hipotiroidi gibi sekonder bir sebep veya başka
bir primer hiperlipidemi gerekir
• Erişkin yaştan önce ortaya çıkmaz
• Avuç içi çizgilerindeki palmar ksantomlar karakteristiktir
• Diz ve dirseklerin ekstansör yüzeylerinde de ksantomlar
vardır
• Koroner arterlerin yanısıra periferik damarları da tutan
prematür ateroskleroz riski artmıştır
Hepatik Lipaz Eksikliği
• HDL fraksiyonu büyük, trigliseritten zengin, HDL2
dansitesindeki partiküllerden oluşmuştur
• Homozigotlarda ciddi hipertrigliseridemi, hiperkolesterolemi
ve artmış IDL konsantrasyonu vardır
• Apo A-I ve HDL-kolesterol konsantrasyonu hafif artmıştır
Kolesterol-7-alfa-hidroksilaz Eksikliği
• Ender görülür
• Safra asidi sentezinin ilk basamağını katalizleyen enzimi
kodlayan gen defektiftir
• Karaciğerde LDLR aktivitesinin azaldığı düşünülmektedir
• Tanı konan az sayıdaki hasta statin tedavisine dirençlidir
Düşük Kolesterol + Düşük Trigliserid
• Abetalipoproteinemi
• Hipobetalipoproteinemi
• Şilomikron Retansiyon Hastalığı
Abetalipoproteinemi
• 1950’de Bassen ve Kornzweig tarafından tarif edilmiş,
1952’de nedeni anlaşılmıştır
• ApoB geni normaldir
• Mikrozomal Trigliserid Transfer Protein (MTP) defektiftir
• ER’da lipidler apo B ile birleşemez
• Apo B içeren lipoproteinler yapılamaz
• Plazmada apo B içeren lipoproteinler ve apo B yoktur
• Yağda eriyen vitaminlerin (özellikle A ve E) emilimi
bozulur
• Trigliserid ve total kolesterol düşüktür
• Hepatosit ve eritrositlerde yağ damlacıkları birikir
Abetalipoproteinemi (Klinik Bulgular)
• Bebeklik çağında büyüme geriliği ile başvuru
• Yağ malabsorpsiyonuna bağlı kronik diyare
• Periferik yaymada akantositoz
• Gözdibinde atipik retinitis pigmentoza
• Gece körlüğü, ataksi
• Şilomikron yokluğu nedeni ile yağda eriyen vitaminlerin
emiliminde bozukluk vardır
• Düşük yağ içeren diyete cevap verir
• Eksik vitaminlerin yerine konması gerekir
Hipobetalipoproteinemi
• ApoB geninde nonsens ve frameshift mutasyonlar sonucu
“budanmış” apo B
• Apo B-100’ün %6.46-%89’u mevcuttur
• Lipid bağlama kapasitesi azalır, %29’un altında lipoprotein
oluşturamaz
• Heterozigotlar genellikle asemptomatiktir
• Bunlarda total kolesterol ve LDL-kolesterol normalin 1/3’ü
kadardır
• Homozigotlarda abetalipoproteinemideki klinik görülür
Şilomikron Retansiyon Hastalığı
• Plazmada apo B-48 bulunmaz
• Şilomikronların enterositteki sekresyon yolunda rolü olan
Sar1 GTPaz proteinin geninde (SARA2) mutasyonlar
saptanmıştır
• Etkilenen kişilerde LDL, HDL ve yağda çözünen vitaminler
düşüktür
• Çocukluk çağında yağ malabsorpsiyonu ve steatore vardır
• Yağda çözünen vitaminler dışarıdan verilmezse nörolojik
disfonksiyon gelişir
• Enterositlerde yağ damlacıkları bulunur
İzole Düşük HDL-kolesterol
• Familyal Hipoalfalipoproteinemi
• Apo A-I eksikliği ve Apo C-III eksikliği
• Apo A-I varyantları
• Tangier hastalığı
• LCAT eksikliği
- Klasik (tam) eksiklik
- Balık gözü hastalığı
Familyal Hipoalfalipoproteinemi
• Oldukça sık görülür (1:400)
• Tanı kriterleri:
- Normal VLDL-kol. Ve LDL-kol. varlığında düşük HDL-kol.
- Sekonder sebeplerin bulunmaması
- Birinci derecede akrabada aynı lipoprotein paterni
• HDL-kol. Erkeklerde < 30 mg/dL
Kadınlarda < 40 mg/dL
Ailelerin yarısında hepatik lipaz veya apo A-I/C-III/A-IV
gen defektleri
Bazı vakalarda ABCA1 gen mutasyonları
Prematür KKH tipiktir
Apo A-I eksikliği ve Apo C-III eksikliği
• 3 tip eksiklik tanımlanmıştır, hepsinde korneada bulanıklık ve
prematür koroner kalp hastalığı görülür
• Sadece tip I’de defekt tanımlanmıştır
• Hem apo A-I hem de apo C-III eksiktir
• Apo A-I geni apo C-III genine komşudur, mutantlarda 5.5
kb’lık bir parça ters yerleşir
• HDL-kol. < 5 mg/dL
Apo A-I varyantları
• En az 11 varyant tanımlanmıştır
• En iyi bilineni olan apo A-IMILANO da HDL- kolesterol
düşüktür ama ateroskleroz insidansı artmamıştır
• Lipid bağlama ile ilgili bölge etkilenmiştir
• Ek olarak LCAT aktivitesi de azalmıştır
Tangier Hastalığı
• Sebebi ABCA1 genindeki mutasyonlardır
• Ender görülür, geçiş otozomal ressesif
• HDL hemen hiç yok, apo A-I ve A-II çok düşük
• HDL yapısal olarak anormaldir
• Makrofajlarda kolesterol esterleri birikir
• Plazma total kolesterol düşük, trigliserid değişir
• Büyük turuncu tonsiller patognomoniktir
• Splenomegali, hafif trombositopeni vardır
• Hepatomegali, lenfadenopati daha az görülür
• Periferik nöropati
• Kardiak risk artmış olmakla birlikte beklenenden
düşüktür (LDL-kolesterol de düşük ve trombositopeni)
LCAT Eksikliği
• Ender görülür, LCAT geninde mutasyon vardır, otozomal
resessiftir
• Serbest kolesterol esterleşemez
• Lipoproteinlerin yapısı ve şekli bozuktur; HDL disk şeklinde
veya küçük küreseldir
• Korneada opasite, hemolitik anemi, proteinüri (böbrek
yetersizliği)
• Plazma trigliseridi yüksek, HDL-kolesterol düşüktür
• Prematür ateroskleroz gelişir
Balık Gözü Hastalığı
Sebebi LCAT geninde mutasyonlardır
Familyal LCAT eksikliğinden farklı olarak tespit edilebilir
LCAT aktivitesi vardır
“Ölü balık gözü” görüntüsüne sebep olan kornea opasitesi
çok düşük HDL düzeyine (normalin %10’u) bağlıdır
LDL trigliseritten zengin, VLDL artmış ama bileşimi
normaldir
İzole Yüksek HDL-kolesterol
• Kolesterol Ester Transfer Protein Eksikliği
Kolesterol Ester Transfer Protein Eksikliği
• CETP genindeki çeşitli mutasyonlar sonucu ortaya çıkar
• Homozigotlarda plazma HDL-kolesterol (> 100 mg/dL), apo
A-I konsantrasyonu yüksektir
• Tamamen yokluğunda HDL-kolesterol normalin 5 katı,
LDL-kolesterol ve apo B normalin %60’ı kadardır
• CETP eksikliği aterom oluşumuna karşı koruyucu olduğundan
CETP’i inhibe eden ilaçlar ile klinik deneyler
sürdürülmektedir
Sekonder Hiperlipidemi Sebepleri
Hiperkolesterolemi (LDL↑)
•
•
•
•
•
•
Hipotiridi
Nefrotik sendrom
Kolestaz
Anoreksia nervosa
İmmünglobulinler
İlaçlar
-Siklosporin
-Sirolimus
-Antiepileptikler
Hipertrigliseridemi (VLDL↑)
DM (tip 1, tip 2)
KBY
Obezite
Alkol
Hipotiroidi
İlaçlar (östrojenler, kortikosteroidler, progestojenler,
retinoidler)
• İmmünglobulinler
•
•
•
•
•
•
Birleşik (Mixed) Hiperlipidemi (VLDL, LDL)
• Hipotiroidi
• Nefrotik sendrom
• İmmünglobulinler
Diabetes mellitus
• Kontrolsüz diyabette hipertrigliseridemi gelişir. Her iki
tipte de insülin eksikliği vardır:
• İnsülin LPL’ı aktive eder fakat yağ dokusunda hormon
duyarlı lipazı inhibe eder
• Diyabette yağ asidi klirensi azalırken karaciğere yağ
asidi akışı arttığından hepatik trigliserid sentezi artar
• Diyabetteki dislipidemi küçük, yoğun LDL ve düşük
HDL-kolesterol ile karakterizedir
Hipotiroidi
• LDL-kolesterol artar çünkü:
• LDL reseptörleri azalır, buna bağlı olarak LDL klirensi
azalır
• LPL aktivitesi de azalabilir
Nefrotik Sendrom
• Hepatik protein sentezi arttığından apo B-100 yapımı da
artar
• LDL-kolesteroldeki artış albuminürinin ciddiyeti ile
ilişkilidir
• HMG-CoA aktivitesi de artmıştır
• LPL aktivitesi azalır: hipertrigliseridemi
Kronik Böbrek Hastalığı
• En sık gözlenen bozukluk hipertrigliseridemidir
• Apo B-48 içeren lipoproteinler ve kalıntıları artmıştır
• Trigliseritten zengin lipoproteinlerin apo C-III içeriği
artmıştır
• Apo C-III hem LPL’ı hem de şilomikron ve VLDL
kalıntılarının karaciğer tarafından alınmasını inhibe eder
• Böbrek yetersizliğinde kardiyovasküler hastalık riskinin
arttığı bilinmektedir
Böbrek Transplantasyonu
• İmmünsupresif ilaçlar hiperlipidemiye sebep olur
• Kortikosteroidler hem hiperkolesterolemi hem de
hipertrigliseridemi yapar
• Takrolimustan çok, sirolimus ve siklosporin ciddi
hiperkolesterolemiye sebep olur
Karaciğer Hastalığı
• Kolesterol ekskresyonunun fizyolojik tek yolu karaciğer
tarafından safraya sekresyondur
• Lipoprotein X kolestaza özgüdür
• Aterojenik potansiyeli tanımlanmamıştır, ancak kolestazda
Lp(a) nın düşük ve HDL-kolesterolün de yüksek olması,
kolestazın kardiyovasküler riski arttıran bir bozukluk olarak
tanınmamasının sebebi olabilir
NASH ve NAFLD
• Non-alkolik steatohepatit (NASH) ve non-alkolik yağlı
karaciğer hastalığı (NAFLD) metabolik sendromun
belirtileri olup kardiyovasküler risk artışı ile ilişkilidir
• Her ikisi tip 2 diyabet, IGT ve IFG ile de ilişkili olabilir
• NASH/NAFLD ile ilişkili dislipidemi ya hipertrigliseridemi
ya da mikst hiperlipidemidir
• Küçük, yoğun LDL partikülleri nedeni ile familyal kombine
hiperlipidemiden ayırdedilmelidir
Alkol
• Yatkın kişilerde hipertrigliseridemi sebebidir
• VLDL’nin yapımı artmış, uzaklaştırılması azalmıştır
• Ağır vakalarda akut pankreatite yol açan şilomikronemi
sendromu ile sonuçlanabilir
• Makul miktarda alınan alkol, HDL-kolesterolü
arttırdığından daha düşük mortalite ile ilişkilidir
Lipid Bozukluklarının Araştırılması
• Serum veya plazmanın görünüşü lipid bozukluğunu
gösterebilir
• Büyük partiküller olan şilomikronlar ve VLDL ışığın
saçılmasına yol açar
• Plazmadan daha az yoğun olan şilomikronlar 4°C’de bir gece
bekletilen örneğin yüzeyinde krema tabakası oluşturur
• VLDL çok yüksek konsantrasyonda ise örneğin bulanık
görünmesine sebep olur
• En az 12 saatlik bir açlıktan sonra alınan örnekte
trigliserid, total ve HDL-kolesterolün ölçülmesi ile tam
lipid profili ortaya çıkar
• Total kolesterol en sık kullanılan ölçüdür, ancak bazı
dislipidemilerde total kolesterol normal düzeydedir. Aç
olmama durumu test sonucunu fazla etkilemez (±3%)
• Trigliserid düzeyi yemekten sonra 2-3 kat artar. Ölçüm
açlıkta yapılmalıdır.
• HDL-kolesterol, çöktürme yöntemi (eski) veya PEG ile
modifiye edilmiş enzimlerin selektif olarak HDL’deki
kolesterol ile reaksiyona girmesi prensibine dayanan
direkt metod ile ölçülür
• LDL-kolesterol, Friedewald formülü ile hesaplanabilir:
LDL-K = TK – (HDL-K + TG/5)
veya direkt metod ile ölçülebilir
• Friedewald formülü ile hesap için açlık durumu gereklidir,
serum trigliserid düzeyi 400 mg/dL’nin altında olmalıdır
• Direkt metodlar için açlık durumu gerekmez
• Apo B ölçümü kardiyovasküler risk ve tedaviye cevap
açısından LDL-kolesterolden daha iyi bir kestiricidir (apo B
ölçümü partikül sayısını gösterir)
LDL-kolesterol ve LDL
Apo B
POLAR
SURFACE COAT
Phospholipid
Free cholesterol
LDL
NONPOLAR
LIPID CORE
Cholesterol Ester
Triglyceride
LDL-kolesterol
James Otvos 2007
LDL partikülleri ateroskleroza
sebep olur
LDL partikül sayısı ne kadar çoksa, ne kadar kolesterol
taşıdıklarına bakmaksızın, kalp krizine sebep olan plak
oluşum riski o kadar artar
Fredrickson et al. NEJM 1967; 276: 148
Non-HDL-kolesterol
Non-HDL-kolesterol = Total kolesterol – HDL-kolesterol
• LDL-kolesterolden farkı VLDL’deki kolesterolü de
içermesidir
• Epidemiyolojik çalışmalar vasküler riski LDL-kolesterolden
daha iyi olarak, hemen hemen apo B kadar, öngördüğünü
göstermiştir
• LDL partikül sayısını yansıtmaktadır
• Ölçümü için açlık durumu gerekmez, apo B testinden
ucuzdur
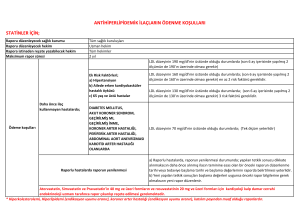
ATP IV’e Göre Statin Tedavisi Gereken 4 Grup
• Kardiyovasküler hastalığı olanlar
• LDL-kolesterolü ≥ 190 mg/dL olanlar
• 40 – 75 yaşındaki tip 2 DM hastaları
• 40 – 75 yaşında olup 10-yıllık kardiyovasküler hastalık
riski ≥ % 7.5 olanlar