CIVANIN KATI FAZ ÖZÜTLEME YÖNTEMİYLE
ZENGİNLEŞTİRİLMESİ, TÜRLEMESİ VE SOĞUK BUHAR
ATOMİK ABSORPSİYON SPEKTROMETRİ YÖNTEMİYLE TAYİNİ
DİLEK ÇABUK
YÜKSEK LİSANS TEZİ
KİMYA
GAZİ ÜNİVERSİTESİ
FEN BİLİMLERİ ENSTİTÜSÜ
EYLÜL 2012
ANKARA
ii
Dilek ÇABUK tarafından hazırlanan CIVANIN KATI FAZ ÖZÜTLEME
YÖNTEMİYLE ZENGİNLEŞTİRİLMESİ, TÜRLEMESİ VE SOĞUK BUHAR
ATOMİK ABSORBSİYON SPEKTROMETRİ YÖNTEMİYLE TAYİNİ adlı bu
tezin Yüksek Lisans tezi olarak uygun olduğunu onaylarım.
Prof. Dr. A. Rehber TÜRKER
Tez danışmanı, Kimya Anabilim Dalı
…………………………..
Bu çalışma, jürimiz tarafından oy birliği ile KİMYA Anabilim Dalında Yüksek
Lisans tezi olarak kabul edilmiştir.
Prof. Dr. Erdoğan HASDEMİR
………………………….
(Kimya Anabilim Dalı, Gazi Üniversitesi)
Prof. Dr. A. Rehber TÜRKER
…………………………..
(Kimya Anabilim Dalı, Gazi Üniversitesi)
Prof. Dr. Orhan ACAR
…………………………...
(Atatürk Meslek Yüksekokulu, Gazi Üniversitesi)
Tarih: 27/09/2012
Bu tez ile G.Ü. Fen Bilimleri Enstitüsü Yönetim Kurulu Yüksek Lisans derecesini
onamıştır.
Prof. Dr. Şeref SAĞIROĞLU
Fen Bilimleri Enstitüsü Müdür
…………………………..
iii
TEZ BİLDİRİMİ
Tez içindeki tüm bilgilerin etik davranış ve akademik kurallar çerçevesinde elde
edilerek sunulduğunu, ayrıca tez yazım kurallarına uygun olarak hazırlanan bu
çalışmada bana ait olmayan her türlü ifade ve bilginin kaynağına eksiksiz atıf
yapıldığını bildiririm.
Dilek ÇABUK
iv
CIVANIN KATI FAZ ÖZÜTLEME YÖNTEMİYLE
ZENGİNLEŞTİRİLMESİ, TÜRLEMESİ VE SOĞUK BUHAR ATOMİK
ABSORPSİYON SPEKTROMETRİ YÖNTEMİYLE TAYİNİ
(Yüksek Lisans Tezi)
Dilek ÇABUK
GAZİ ÜNİVERSİTESİ
FEN BİLİMLERİ ENSTİTÜSÜ
Eylül 2012
ÖZET
Bu çalışmada, Hg(II) ve MeHg iyonları adsorban olarak Amberlyst 36
kullanılarak kolon katı faz özütleme yöntemiyle zenginleştirildi. Cıva türleri,
MHS 15 soğuk buhar sistemi takılı soğuk buhar atomik absorpsiyon
spektrometri yöntemi ile tayin edildi. Her iki cıva türü için de ayrı ayrı en
uygun tutunma ve geri alma değişkenleri belirlendi. Bu amaçla, pH’nın, örnek
çözeltisi akış hızı ve hacminin, geri alma çözeltisinin tipi ve derişiminin cıva
türlerinin geri kazanım verimine etkisi araştırıldı. Yabancı iyonların etkileri de
çalışıldı. Hg(II) ve MeHg iyonlarının her ikisi için de en uygun pH, örnek
çözeltisi akış hızı ve hacmi sırasıyla, 4,0, 3 mL/min ve 50 mL olarak bulundu.
Her iki tür de pH 4’te tutunmasına karşın, iyonların geri alınması farklı geri
alma çözeltileriyle gerçekleştirilebildi. Türleme, bu özellikten yararlanılarak
başarıldı. MeHg iyonları 10 mL 0,1 mol/L HCl çözeltisiyle geri alınırken, Hg(II)
iyonları 3 mol/L HCl içinde 10 mL 0,2 mol/L tiyoüre çözeltisiyle geri alındı.
Belirlenen en uygun deney koşulları altında Hg(II) ve MeHg için gözlenebilme
sınırları sırasıyla 0,44 µg/L ve 0,56 µg/L olarak bulundu. Geri alma çözeltisi
hacmi 10 mL ve örnek hacmi 50 mL olduğundan zenginleştirme faktörü
yalnızca 5 olarak elde edildi. Önerilen türleme ve zenginleştirme yönteminin
doğruluğunu kontrol etmek için belgeli referans madde (ERM-CE464 Tuna
Fish) analiz edildi ve MeHg %7,8 bağıl hata ile tayin edilebildi. Belgeli referans
v
maddedeki Hg(II) derişimi yöntemin gözlenebilme sınırının altında olduğundan
tayin edilemedi. Bununla birlikte, doğruluk, her iki tür ve toplam cıva için
katkılı su ve balık örnekleri analiz edilerek de kontrol edildi. Katkılı örnek
analizlerinde de bağıl hata %10’dan düşük bulundu. Her iki tür için de geri
kazanma veriminin bağıl standart sapması, tekrarlanan beş tayin için %3’ün
altında bulundu.
Bilim Kodu
: 201.1.004
Anahtar Kelimeler : Zenginleştirme, türleme, cıva, adsorpsiyon,
katı faz özütleme, atomik absorpsiyon spektrometri
Sayfa Adedi
: 88
Tez Yöneticisi
: Prof. Dr. Ali Rehber TÜRKER
vi
PRECONCENTRATION AND SPECIATION OF MERCURY BY
SOLID PHASE EXTRACTION METHOD AND DETERMINATION BY
COLD VAPOR ATOMIC ABSORPTION SPECTROMETRIC METHOD
(M. Sc. Thesis)
Dilek ÇABUK
GAZI UNIVERSITY
INSTITUTE OF SCIENCE AND TECHNOLOGY
September 2012
ABSTRACT
In this study, Hg(II) and MeHg ions were preconcentrated by column solid
phase extraction method using Amberlyst-36 as an adsorbent. Mercury species
were determined by cold vapor atomic absorption spectrometry method
mounted with MHS 15 cold vapor system. Optimum retention and elution
parameters were determined for both mercury species separately. For this
purpose, the effect of pH, flow rate and volume of sample solution, type and
concentration of eluent on the recovery of mercury species were investigated.
Effect of foreign ions was also studied. Optimum values of pH, flow rate and
volume of sample solution were found as 4.0, 3 mL/min and 50 mL, respectively
for both Hg(II) and MeHg ions. Although the both species were retained at pH
4, the elution of them could be performed only with different eluents. The
speciation was achieved by using this behavior. While MeHg ions were eluted
with 10 mL of 0.1 mol/L HCl solution, Hg(II) ions were eluted with 10 mL of 0.2
mol/L thiourea solution in 3 mol/L HCl. Under the optimum experimental
conditions determined, limit of detections for Hg(II) and for MeHg were found
as 0.44 µg/L and 0.56 µg/L, respectively. Because of the eluent volume is 10 mL
and the sample volume is 50 mL, only 5 were obtained as preconcentration
factor. To check the accuracy of the proposed speciation and preconcentration
method certified reference material (ERM-CE464 Tuna Fish) was analyzed and
MeHg could be determined with a relative error of 7.8%. Because Hg(II)
vii
concentration in certified reference material is lower than the detection limit of
the method, it could not be determined. However, the accuracy was also checked
by analyzing spiked water and fish samples for both species and total mercury.
Relative error for spiked samples was also found below 10%. Relative standard
deviation of recovery for both species was found below 3% for five replicates.
Science Code : 201.1.004
Key Words
: Preconcentration, speciation, mercury, adsorption,
solid-phase extraction, atomic absorption spectrometry
Page Number : 88
Adviser
: Prof. Dr. Ali Rehber TÜRKER
viii
TEŞEKKÜR
Çalışmam boyunca benden, değerli vaktini, engin bilgi birikimini ve tecrübelerini
esirgemeyen, danışman hocam Sayın Prof. Dr. Ali Rehber TÜRKER’e,
Tez çalışmam boyunca bilgi ve tecrübeleri ile bana yardımcı olan hocalarım Sayın
Prof. Dr. Adalet TUNÇELİ ve Sayın Prof. Dr. Orhan ACAR’a,
Aynı laboratuvarda çalıştığım desteklerini ve bilgilerini hiçbir zaman esirgemeyen
değerli hocalarım Sayın Araş. Gör. Dr. Özcan YALÇINKAYA’ya ve Sayın Araş.
Gör. Hakan Erdoğan’a,
Tez çalışmalarım süresince hep yanımda olup beni destekleyen bütün arkadaşlarıma,
Her zaman yanımda olan sevgisini, maddi ve manevi desteğini bir an olsun
esirgemeyen annem Kudret ÇABUK’a ve babam Teyfik ÇABUK’a,
Teşekkürü bir borç bilirim.
ix
İÇİNDEKİLER
Sayfa
ÖZET........................................................................................................................... iv
ABSTRACT ................................................................................................................ vi
TEŞEKKÜR..............................................................................................................viii
İÇİNDEKİLER............................................................................................................ix
ÇİZELGELERİN LİSTESİ ....................................................................................... .xv
ŞEKİLLERİN LİSTESİ............................................................................................xvi
SİMGELER VE KISALTMALAR..........................................................................xvii
1. GİRİŞ………………………………………………………………………………1
2. GENEL BİLGİLER .................................................................................................3
2.1. Eser Elementlerin Zenginleştirilmesi, Türlemesi ve Tayini .............................3
2.1.1. Eser elementler .......................................................................................3
2.1.2. Eser elementlerin zenginleştirilmesi.......................................................4
2.1.3. Katı faz özütleme yöntemi......................................................................5
2.1.4. Eser elementlerin türlemesi.....................................................................9
2.2. Katı faz özütleme yöntemi ile ilgili bazı çalışmalar........................................10
2.2.1. Katı faz özütleme yöntemiyle yapılan
zenginleştirme çalışmaları......................................................................11
2.2.2. Literatürde yeralan bazı cıva türlemesi çalışmaları…….....................13
2.3. Atomik Absorpsiyon Spektroskopisi (AAS) Yöntemi...................................17
2.3.1. Genel bilgiler........................................................................................17
2.3.2. Atomik absorpsiyon spektrometreleri...................................................20
x
Sayfa
2.3.3. Işın kaynakları.......................................................................................21
2.3.4. Atomlaştırıcılar ....................................................................................25
2.3.5. Monokromatör......................................................................................33
2.3.6. Dedektör ...............................................................................................34
2.4. Atomik Absorpsion Spektroskopisinde Girişimler ........................................34
2.4.1. Fiziksel girişimler ................................................................................35
2.4.2. Kimyasal girişimler ..............................................................................35
2.4.3. İyonlaşma girişimi ................................................................................36
2.4.4. Spektral girişimler ................................................................................37
2.4.5. Zemin girişimi ......................................................................................37
2.5. AAS Yöntemi ile Nicel Tayin ........................................................................39
2.5.1. Kalibrasyon yöntemi ............................................................................40
2.5.2. Standart ekleme yöntemi ......................................................................40
2.6. Analitik Başarımı ile İlgili Terimler ..............................................................41
2.6.1. Duyarlık ...............................................................................................41
2.6.2. Doğruluk ..............................................................................................42
2.6.3. Kesinlik ................................................................................................42
2.6.4. Gözlenebilme sınırı ..............................................................................42
2.6.5. Tayin sınırı ...........................................................................................44
3. DENEYSEL KISIM ...............................................................................................45
3.1. Cihazlar ve Malzemeler...................................................................................45
3.1.1. Atomik absorpsiyon spektrometresi......................................................45
3.1.2. pH metre................................................................................................45
xi
Sayfa
3.1.3. Çalkalamalı su banyosu........................................................................45
3.1.4. Cam kolonlar.........................................................................................46
3.2. Reaktifler, Çözeltiler ve Kimyasal Maddeler.................................................46
3.2.1. Amberlyst 36….....................................................................................46
3.2.2. Cıva (II) stok çözeltisi, 1000 mg/L ….................................................46
3.2.3. Metil cıva stok çözeltisi, 1000 mg/L.. ..................................................46
3.2.4. Kobalt (II) stok çözeltisi, 1000 mg/L. ..................................................46
3.2.5. Nikel (II) stok çözeltisi, 1000 mg/L .....................................................47
3.2.6. Sodyum stok çözeltisi, 1000 mg/L .......................................................47
3.2.6. Potasyum stok çözeltisi, 1000 mg/L ....................................................47
3.2.8. Mangan (II) stok çözeltisi, 1000 mg/L….............................................47
3.2.9. Magnezyum stok çözeltisi, 1000 mg/L……………………………….47
3.2.10. Çinko stok çözeltisi, 1000 mg/L’lik ...................................................47
3.2.11. Kalsiyum stok çözeltisi, 1000 mg/L ..................................................48
3.2.12. Bakır (II) stok çözeltisi, 1000 mg/L ...................................................48
3.2.13. Amonyak çözeltisi, 0,1 mol/L’lik ......................................................48
3.2.14. Hidroklorik asit çözeltisi, 3,0 mol/L ..................................................48
3.2.15. Hidroklorik asit çözeltisi, 2,0 mol/L .................................................48
3.2.16. Hidroklorik asit çözeltisi, 1,0 mol/L ..................................................48
3.2.17. Hidroklorik asit çözeltisi, 0,5 mol/L ..................................................49
3.2.18. Hidroklorik asit çözeltisi, 0,1 mol/L ..................................................49
3.2.19. Nitrik asit çözeltisi, 3,0 mol/L ............................................................49
3.2.20. Nitrik asit çözeltisi, 2,0 mol/L ............................................................49
xii
Sayfa
3.2.21. KMnO4 çözeltisi, 0,1 mol/L. ..............................................................49
3.2.22. NaBH4 çözeltisi, % 0,5 (m/V)’lik
(%0,5 (m/V)’lik NaOH çözeltisinde) .................................................49
3.2.23 Tiyoüre (3 M HCl çözeltisinde), 0,05 M .............................................50
3.2.24. Tiyoüre (3 M HCl çözeltisinde), 0,1 M ..............................................50
3.2.25. Tiyoüre (1 M HCl çözeltisinde), 0,2 M……………………………..50
3.2.26. Tiyoüre (2 M HCl çözeltisinde), 0,2 M……………………………..50
3.2.27. Tiyoüre (3 M HCl çözeltisinde), 0,2 M……………………………..50
3.2.28. Arsenik(III) stok çözeltisi, 1000 mg/L...............................................50
3.2.29. Selenyum(IV) stok çözeltisi, 1000 mg/L............................................51
3.3. Örneklerin Hazırlanması…………………………………………………….51
3.3.1. Model çözeltiler………………………………………………………51
3.3.2. Su örneklerinin hazırlanması…………………………………………51
3.3.3. Balık örneğinin hazırlanması…………………………………………52
3.3.4. Belgeli referans maddelerin hazırlanması…………………………….52
3.4. Katı Faz Özütleme (SPE) ile Genel Zenginleştirme Yöntemi………………52
3.4.1. Kolonun hazırlanması ..........................................................................52
3.4.2. Zenginleştirme, türleme ve tayin yöntemi ...........................................53
3.4.3. Adsorpsiyon kapasitesi ve tayin yöntemi .............................................54
4. SONUÇLAR .........................................................................................................56
4.1. Cıva Türlemesi ve Tayini ...............................................................................56
4.1.1. CVAAS yöntemi ile cıva türlemesi ve tayini için en uygun
yöntem ve değişkenleri……………………………………………….56
xiii
Sayfa
4.1.2. Yöntemin analitik başarımı...…………………………………………....67
4.1.3. Model çözeltide cıva tayini ve türlemesi……………………………......70
4.1.4. Su örneklerinde cıva tayini ve türlemesi………………………………..70
4.1.5. Balık örneğinde cıva tayini ve türlemesi……………………………….70
4.1.6. Adsorpsiyon izotermi ve adsorpsiyon kapasitesi………………………..74
5. TARTIŞMA……………………………………………………………………...76
KAYNAKLAR……………………………………………………………………..82
ÖZGEÇMİŞ………………………………………………………………………...88
xiv
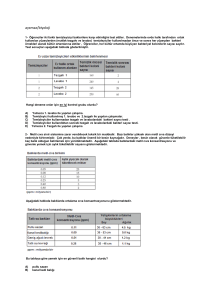
ÇİZELGELERİN LİSTESİ
Çizelge
Sayfa
Çizelge 2.1. Bazı alev türlerinin özellikleri…………………………………………25
Çizelge 3.1. CVAAS yöntemiyle Hg tayininde aletsel değişkenler ………………..45
Çizelge 3.2. Su örneklerinin alındıkları andaki pH değerleri ve sıcaklıkları………..51
Çizelge 4.1. Geri alma çözeltisi cinsi ve derişiminin Hg(II)’ nin geri kazanma
verimine etkisi………………………………………………………...59
Çizelge 4.2. Geri alma çözeltisi cinsi ve derişiminin MeHg ‘nın geri kazanma
verimine etkisi…………………………………………………………59
Çizelge 4.3. Hg(II)’ nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi………………………………………………...62
Çizelge 4.4. MeHg’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi………………………………………………...64
Çizelge 4.5. Hg(II) ve MeHg’nin Amberlyst 36 dolgulu kolonda
zenginleştirilmesi ve türlemesi için bulunan analitik
başarım ölçütleri…………………………………………………….....69
Çizelge 4.6. Model çözelti ile cıva türleme çalışması………………………………71
Çizelge 4.7. Su örneklerindeki cıva türleri tayini…………………………………..72
Çizelge 4.8. Balık örneğindeki cıva türleri tayini…………………………………..73
Çizelge 4.9. Cıvanın Amberlyst 36 ile zenginleştirilmesi ve türlemesi
çalışmasının literatürdeki diğer çalışmalarla karşılaştırılması………...81
xv
ŞEKİLLERİN LİSTESİ
Şekil
Sayfa
Şekil 2.1. Kolon tekniği ile zenginleştirme işlemi......................................................8
Şekil 2.2. Atomik absorpsiyon spektrometresinin şematik gösterilişi .................... .21
Şekil 2.3. Oyuk katot lambasının şematik gösterilişi .............................................. .23
Şekil 2.4. Ön-karıştırmasız alev başlığının şematik görünüşü ................................ .26
Şekil 2.5. Ön-karıştırmalı alev başlığının şematik görünüşü .................................. .27
Şekil 2.6. MX çözeltisinin alevde atomlaşması ve uyarılması için olasılıklar........ .28
Şekil 2.7. Grafit fırınlı atomlaştırıcının şematik görünüşü ..................................... .30
Şekil 2.8. CVAAS yönteminin şematik gösterilişi………………………………….33
Şekil 2.9. Standart ekleme kalibrasyon grafiği örneği ............................................ .41
Şekil 3.1. Katı faz özütleme tekniğinde kullanılan kolonun şematik görüntüsü ..... .53
Şekil 4.1. Geri alma çözeltisi olarak 0,1 mol/L HCl çözeltisi kullanıldığında
pH’nın geri kazanma verimine etkisi……………………………………57
Şekil 4.2. pH’ nın cıva türlerinin geri kazanma verimine etkisi……………………58
Şekil 4.3. Akış hızının cıva türlerinin geri kazanma verimine etkisi……………….60
Şekil.4.4. Hacmin cıva türlerinin geri kazanma verimine etkisi……………………61
Şekil 4.5. Hg(II) için kalibrasyon grafiği…………………………………………...68
Şekil 4.6. MeHg için kalibrasyon grafiği…………………………………………...68
Şekil 4.7. Hg(II) için doğrusallaştırılmış Langmuir eşitliğinin grafiği……………..74
Şekil 4.8. MeHg için doğrusallaştırılmış Langmuir eşitliğinin grafiği……………..75
xvi
SİMGELER VE KISALTMALAR
Bu çalışmada kullanılmış bazı simgeler ve kısaltmalar, açıklamaları ile birlikte
aşağıda sunulmuştur.
Simgeler
Açıklama
b
Langmiur sabiti
Ce
Denge derişimi
N
Ölçüm sayısı
Q0
Langmuir Adsorpsiyon kapasitesi
Kısaltmalar
Açıklama
AAS
Atomik Absorpsiyon Spektroskopisi
AFS
Atomik Floresans Spektroskopisi
CVAAS
Soğuk Buhar Atomik Absorpsiyon
Spektroskopisi
ETAAS
Elektrotermal Atomik Absorpsiyon
Spektroskopisi
FAAS
Alevli Atomik Absorpsiyon Spektroskopisi
HGAAS
Hidrür Oluşturmalı Atomik Absorpsiyon
Spektroskopisi
ICP-OES
İndüktif Eşleşmiş Plazma-Optik Emisyon
Spektroskopisi
ICP-MS
İndüktif Eşleşmiş Plazma-Kütle Spektroskopisi
MeHg
Metil cıva iyonu (CH3Hg+)
SPE
Katı Faz Özütleme
1
1. GİRİŞ
Canlı metabolizması, canlıda bulunan eser elementlerin az veya çok miktarda
olmasından etkilenmektedir. Eser elementlerin canlı organizmalar içerisindeki
miktarları
çevre
koşulları
ve
besin
türlerine
göre
değişmektedir.
Canlı
organizmalarda herhangi bir işlevin yerine getirilebilmesi için organizmada eser
elementlerin belirli miktarlarda olması gerekir. Bu nedenle, canlıların doğrudan veya
dolaylı olarak temas halinde bulunduğu su, gıda, doku, çevre gibi ortamlardaki eser
elementler sürekli tayin edilmeli ve kontrol edilmelidir [Oliveira ve ark., 2000; Chen
ve Teo, 2001; Grindlay ve ark., 2011].
Genellikle alkali metaller dışındaki metal iyonları su, gıda, çevre gibi örneklerde
düşük derişimlerde yani eser miktarda bulunurlar. Eser elementlerin yüksek doğruluk
ve kesinlikle tayini aşağıdaki nedenlerden dolayı oldukça zordur: (1) Derişimlerin
doğrudan tayininin yapılamayacak kadar küçük olması, (2) Örneğin fiziksel halinin
tayin yöntemine uygun olmaması, (3) Ortamda bulunan diğer element veya
bileşiklerin bozucu etkileri olması. Bu nedenle, belirtilen zorlukların giderilmesi ve
bilinen yaygın tayin yöntemlerinin kullanım alanlarının genişletilmesi için tayin
öncesi örnek üzerinde çeşitli çalışmalar yapılmaktadır [Welz ve Sperling, 1999; Dos
Santos ve ark., 2005].
Tayin öncesi örnek üzerinde yapılan ön işlemler, alet geliştirmeye göre daha basit ve
daha az maliyet gerektirdiğinden bu konuda daha fazla çalışma yapılmaktadır. Tayin
öncesi yapılan ön işlemler, örneğin analize hazırlanması, çözülmesi, girişimlerin
azaltılması veya giderilmesi için bileşenlerin birbirinden ayrılması, tayin sınırını
düşürmek için eser elementlerin deriştirilmesi ve tayin için uygun ortama alınması
gibi işlemleri kapsar.
Eser elementlerin başka bir ortama alınarak daha küçük hacimde toplanması işlemine
“zenginleştirme” denir. Zenginleştirme yapılırken aynı zamanda bir ayırma işlemi de
yapılmaktadır. Eser düzeydeki elementlerin ayrılması ve/veya zenginleştirilmesinde
sıvı-sıvı özütleme, iyon değiştirme, adsorpsiyon (katı faz özütleme), flotasyonla
2
zenginleştirme, bulutlanma noktası özütlemesi, elektrolitik biriktirme, birlikte
çöktürme ve uçurma ile zenginleştirme teknikleri kullanılır. Ayırma ve
zenginleştirme, tıp, tarım, gıda, eczacılık, çevre kirliliği, savunma ve birçok sanayi
alanında doğru tayinlerin yapılabilmesi için en çok uygulanan analitik ön işlemlerden
biridir. Zenginleştirme ve/veya ayırmanın temel amacı, düşük derişimdeki eser
metalleri karmaşık örnek ortamlarından ayırmak, bilinen bir ortama almak ve bu
arada deriştirerek tayin yöntemi için girişimlerin olmadığı bir ortamda duyarlı olarak
tayin etmektir [Kalfa, 2009; Lemos ve ark., 2008; Zhang ve ark., 2008].
Türleme genel olarak örneklerde tayin edilen türün kimyasal şeklinin (iyon, molekül
vb.) belirlenmesi ve tayin edilmesi olarak tanımlanır. Elementlerin zehirlilikleri
kimyasal türleri ile yakından ilişkilidir ve bu nedenle elementin örnekte hangi türü
halinde bulunduğunun belirlenmesi analitik kimyada oldukça önemlidir. Ayrıca
metallerin çevrede taşınması, bitkiler ve hayvanlar tarafından alınması ve
depolanması bunların kimyasal türlerine bağlı olarak değişmektedir.
Bu tez kapsamında katı-faz özütlemesi yöntemiyle Hg(II) ve CH3Hg+ (MeHg)
iyonlarının Amberlyst 36 dolgulu kolonda zenginleştirilmesi ve türlemesi
amaçlanmıştır. Zenginleştirme ve türleme çalışmasında kolon yöntemi kullanılmış ve
bu yöntem için pH, örnek çözelti akış hızı, örnek çözeltisi hacmi, geri alma çözeltisi
cinsi ve derişimi gibi faktörlerin etkisi incelenmiştir. Belirlenen en uygun deneysel
koşullarda örnek çözeltileri soğuk buhar
oluşturmalı
atomik absorpsiyon
spektrometresi (CVAAS) yöntemiyle analiz edilmiştir. Tayinin doğruluğu ve
kesinliği de araştırılmıştır.
3
2. GENEL BİLGİLER
2.1. Eser Elementlerin Zenginleştirilmesi, Türlemesi ve Tayini
2.1.1. Eser elementler
Bir örnekte, örneği oluşturan diğer bileşenlere göre çok az bulunan elementlere “eser
element” denir. Günümüzde % 10-2 – 10-6 derişim aralığı eser derişim, % 10-6’nın
altındaki derişimler ise ultra-eser derişim olarak kabul edilmektedir. Eser bileşenler
dışında % 1-5 arasında değişen bileşenlere yan bileşenler, % 5 ve daha yüksek
derişimdeki bileşenlere de ana bileşen denir. Eser elementlerin tayininde atomik
absorpsiyon spektrometrisi, atomik emisyon spektrometrisi, nötron aktivasyon
analizi, elektroanalitik yöntemler, X ışınları floresans spektrometresi, atomik
floresans spektrometrisi ve kütle spektrometri gibi yöntemler kullanılmaktadır.
Günümüzde litrede miligram ve mikrogram mertebesinde elementler yukarıda sözü
edilen uygun analitik yöntemler ile yüksek doğruluk ve güvenilirlikte tayin
edilebilmektedir. Daha düşük düşük derişimlerde yöntemin tayin sınırının altında
kalındığı için duyarlı ve tekrarlanabilirliği yüksek ölçümler yapılamamaktadır.
Ayrıca, birçok ortamda bu derişimdeki elementlerin tayini örnek ortamının fiziksel
ve kimyasal çevresinden dolayı ortaya çıkan girişimler nedeniyle oldukça zor
yapılmaktadır. Girişim etkisinin olmadığı veya giderildiği ortamlar, eser element
tayini için uygun ortamlardır. Aletli analiz yöntemleri ile eser element tayinlerinde
girişim etkilerinin olmaması için standartlar ile örneklerin fiziksel ve kimyasal
özelliklerinin mümkün olduğu kadar birbirine benzetilmesi gerekmektedir.
Bu problemlerin giderilmesi ve tayin elementini gerek uygun bir ortama almak,
gerekse deriştirmek amacı ile ayırma-zenginleştirme yöntemleri olarak adlandırılan
çeşitli örnek ön işlemleri geliştirilmiştir. Bu yöntemlere genel olarak zenginleştirme
yöntemleri denilmektedir.
4
2.1.2. Eser elementlerin zenginleştirilmesi
Karmaşık ortamlarda girişim yapabilecek türlerin çokluğu ve tayin elementinin
miktarının eser seviyede olmasından dolayı genellikle sağlıklı bir analiz mümkün
değildir. Bu nedenle, eser element tayini yapılmadan önce bir ayırma-zenginleştirme
basamağının uygulanması zorunlu hale gelir. Zenginleştirme işlemleri ile tayin
edilecek madde hem tayin tekniğine uygun olan bir ortama alınır hem de daha küçük
hacim içerisinde toplanarak deriştirilir.
Ayırma, bir maddenin temasta bulunan iki faz arasında değişik oranda dağılması
esasına dayanır. Bütün ayırma yöntemlerinde katı-sıvı, sıvı-sıvı, sıvı-gaz ve katı-gaz
şeklinde olabilen iki faz bulunmaktadır.
Genel olarak eser element çalışmalarında, eser bileşenler katı veya çözülmüş
örneklerden ayrılırken, ana bileşen çözeltide kalır. Ana bileşen örnekten
uzaklaştırılırken, eser bileşenlerin çözeltide kaldığı uygulama şekli eser element
tayinlerinde pek kullanılmaz. Çünkü ana bileşen çözeltiden ayrılırken beraberinde
eser elementleri de sürükleyebilir. Eser elementlerin birbiri üzerine girişimleri
(spektral girişim gibi) varsa, eser bileşenlerin diğer eser bileşenlerden ayrılması
gerekebilir.
Eser element tayinlerinde kullanılan ayırma-zenginleştirme yöntemleri ile tayin
basamağında sağlanan iyileştirmeler şunlardır:
1. Eser element derişimi artırılarak yöntemin duyarlığı artırılır.
2. Eser elementler bilinen ve uygun bir ortama alındığından, ortamdan gelebilecek
girişimler giderilir.
3. Büyük örnek hacimleri ile çalışılabildiği için örneğin homojen olmayışından
gelebilecek hatalar önlenir.
4. Ayırma işlemi ile eser elementler bilinen ortam içine alındığından, standartlar ile
örnek ortamını benzetmek kolaylaşır.
5
Zenginleştirme basamağının esas amacı, analit için gözlenebilme sınırını düşürmek,
girişimlerden kurtulmak ve tayin edilecek türün derişimini tayin edilebilecek düzeye
artırmaktır. Derişimin ne kadar artırıldığı zenginleştirme faktörüne bağlıdır.
Zenginleştirme faktörü, tayin edilecek türün orijinal örneğe göre kaç kez daha derişik
hale getirildiğinin ölçüsüdür.
Zenginleştirmede önemli olan bir başka değişken de geri kazanma verimidir. İdeal
bir ayırmada ayrılmak istenen türün tamamı bulunduğu ortamdan ayrılır. Bu
durumda geri kazanma verimi % 100 olur. Fakat uygulamada % 99’dan daha iyi geri
kazanma verimine ulaşmak genellikle mümkün değildir. Düşük derişimlerde
çalışıldığında, % 90 veya % 95’lik geri kazanma verimleri analitik amaçlar için
yeterli kabul edilmektedir [Mizuike, 1983].
Eser element zenginleştirme yöntemlerinden yaygın olarak kullanılanların adları
aşağıda sıralanmıştır. Bu çalışmada katı faz özütleme tekniği kullanıldığından sadece
bu teknik hakkında ayrıntılı bilgi verilmiştir.
1. Sıvı-sıvı özütleme yöntemi
2. Elektroliz ile biriktirme yöntemi
3. Uçuculaştırma yöntemi
4. İyon değiştirme yöntemi
5. Birlikte çöktürme yöntemi
6. Flotasyon yöntemi
7. Katı faz özütleme yöntemi
2.1.3. Katı faz özütleme yöntemi
Katı faz özütleme yöntemi, sıvı faz içerisinde bulunan tayin edilecek türün katı bir
faz üzerinde adsorbe olmasına dayanan bir yöntemdir. Zenginleştirme yöntemleri
arasında katı faz özütleme yöntemi, sıvı-sıvı özütleme yöntemi gibi diğer
zenginleştirme yöntemlerine göre basit, hızlı, ucuz olması ve yüksek zenginleştirme
6
faktörü elde edilebilmesinden dolayı daha çok tercih edilir. Bu yöntemin sıvı-sıvı
özütlemesine göre birçok üstünlükleri vardır. Bu üstünlükler şunlardır;
Katı faz özütlemesi kolonda yapıldığında, analizi yapılacak örnek çözeltisi
katı faz özütleme kolonu içinden hızlıca geçebilir. Tutunan türler bir organik
çözücü veya bir başka uygun geri alma çözeltisinin küçük bir hacmi ile hızla
kolondan alınabilir. Bunun aksine, basit sıvı-sıvı özütleme yöntemi, özütleme
sıvısının eklenmesi, çalkalanması, emülsiyonun ayrılmasının beklenmesi ve
iki sıvı fazın dikkatlice ayrılması gibi elle yapılan önemli ve uzun işlem
basamakları içerir.
Katı faz özütleme yönteminde geri alma çözeltisi olarak inorganik
(çoğunlukla) ve organik çözücüler kullanılır. Geri alma çözeltisi olarak
kullanılan çözelti miktarı genellikle 10 mL’yi aşmaz. Analitik ayırmalarda
organik çözücülerin büyük miktarlarının kullanılması önemli çevre sorunları
doğurur. Ayrıca, organik çözücüler nispeten büyük hacimlerde kullanıldığı
zaman, tayin edilecek türün kirlenme riski artar.
Katı faz özütleme yönteminin diğer bir üstünlüğü katı fazın tekrar tekrar
kullanılabilmesidir. Halbuki sıvı-sıvı özütlemesinde sıvı faz her defasında
yenilenmelidir.
Katı faz özütleme yöntemi yüksek zenginleştirme faktörüne sahiptir.
Katı faz özütleme işlemleri, akışa enjeksiyon tekniği ile kolaylıkla
birleştirilebilmektedir. Bu nedenle zenginleştirme tekniklerinde önemli
kolaylıklar ve üstünlükler sağlamaktadır
Zenginleştirme ve ayırma işlemlerinde katı faz özütleme yöntemi genelde üç farklı
şekilde uygulanır. Bunlar, kolon tekniği, çalkalama tekniği ve yarı geçirgen süzgeç
(membran) ile süzme tekniğidir.
7
Bu bahsedilen üç teknikten hangisi kullanılırsa kullanılsın, katı faz özütleme yöntemi
dört temel işlem basamağını içerir [Poole ve Poole, 1997].
Şartlandırma basamağı; Katı faz ile örnek temas ettirilmeden önce, katı faz pH,
iyonik şiddet, polarite gibi özellikler yönünden örnek çözücüsüne benzer bir
çözeltinin (tanık çözelti) temas ettirilmesi ile şartlandırılır. Bu basamağın eksik veya
yetersiz uygulanması genellikle tayin edilecek türün zayıf alıkonması ile sonuçlanır.
Yükleme basamağı; Tayin edilecek türü içeren örnek çözeltisinin katı faz ile temas
ettirilmesi ile analitin katı faz üzerinde adsorplanması sağlanır.
Yıkama basamağı; Diğer bileşenlerin adsorbandan veya katı fazdan uzaklaştırılması
için katı faz bir çözücü ile muamele edilir. Bu basamak için çözücünün seçimi
önemlidir. Çözücü tayin edilecek türü etkilemeksizin diğer bileşenleri önemli ölçüde
katı fazdan sökebilmelidir.
Geri alma basamağı; Tayin edilecek türleri katı fazdan sökmek için katı faz, küçük
hacimde bir çözelti veya bir saf çözücü ile temas ettirilir. Bu basamakta geri alma
çözücüsünün tayin edilecek maddeyi tamamen katı fazdan sökmesi beklenir
[Erdoğan, 2011].
Yükleme ve/veya geri alma basamağındaki başarısızlık geri kazanma veriminin
düşmesine yol açar. Yüksek geri kazanma verimi için hem yükleme hem de geri
alma tam olmalıdır.
Kolon tekniği
Katı faz özütleme tekniği özellikle kolon sistemine uygulandığında oldukça basit ve
hızlı olabilmektedir. Şekil 2.1’de şematik olarak çalışma yönteminin verildiği bu
teknik katı faz özütleme yönteminde en yaygın kullanılan tekniktir ve sürekli sistem
olarak bilinir. Eser element zenginleştirmelerinde genellikle 100–500 mg
adsorplayıcı içeren mini kolonlar kullanılır. Bazı katı faz özütleme tekniklerinde
8
kullanılan kolonlar camdan yapılmış musluklu bir boru olup, musluğun üstünde katı
faz tutucusu gözenekli disk veya cam pamuğu desteği bulunur. Bu destek üzerine
sabit faz olarak katı madde yerleştirilir. Genellikle sabit faz olarak kullanılacak bu
katının üzerine de cam pamuğu desteği konulur. Sabit fazın kolonda iyice yerleşmesi
hareketli sıvı fazın kolondan geçmesi ile sağlanır. Kolon dolgu maddesi tanık çözelti
ile şartlandırıldıktan sonra hazırlanan örnek çözeltisi kolondan belirli bir akış hızı ile
geçirilir. Adsorbanda tutunan tayin elementlerini geri almak için kolona az miktarda
(5-10 mL) çözücü (geri alma çözeltisi) ilave edilir ve bir ölçülü balonda toplanır.
Kolon dolgu maddesinden (adsorban), geri alma çözücüsüne alınan tayin elementleri
uygun bir analiz yöntemi ile tayin edilir. Böylelikle 1000 kata varan
zenginleştirmeler yapılabilir.
Şekil 2.1. Kolon tekniği ile zenginleştirme işleminin şematik gösterilişi [Erdoğan,
2011]
Çalkalama tekniği
Tayin edilecek elementin içinde bulunduğu çözeltiye, katı faz maddesi (adsorban)
katılarak belirli süre birlikte çalkalanır. Çalkalama mekanik veya ultrasonik olarak
9
yapılabilir. Tutunma dengesi kurulduktan sonra, katı faz, süzme veya dekantasyon ile
çözeltiden ayrılır. Daha sonra katı faz uygun bir çözücü ile muamele edilerek katı
fazda tutunan elementlerin çözücüye geçmesi sağlanır. Çözücü belirli bir hacme
tamamlandıktan sonra içerdiği elementler tayin edilir. Bazı durumlarda katı fazdaki
eser elementler, katı fazdan geri alınmadan doğrudan da tayin edilebilir. Bu tür
tayinlerde adsorban uygun bir çözücü ile çözelti haline dönüştürülebilir veya katı faz
bulamaç haline getirilerek de doğrudan analiz edilebilir.
Yarı geçirgen süzgeç (membran) ile süzme tekniği
Yarı geçirgen süzgeç ile süzme tekniğinde örnek çözeltisi, çözeltideki türleri tutma
özelliğine sahip bir yarı geçirgen süzgeçten süzülür. Süzgeçte tutunan elementler
uygun bir geri alma çözeltisi ile geri alınır ve tayin edilir. Bu teknik, daha çok
dağılma katsayısının ve tutunma hızının büyük olduğu durumlarda uygulanır.
2.1.4. Eser elementlerin türlemesi
Elementlerin çoğu örnek ortamında birden fazla kimyasal yapıda bulunabilirler. Bazı
elementler farklı yükseltgenme basamakları halinde inorganik bileşikleri halinde
veya organik bileşikleri halinde bulunabilmektedir. Farklı yükseltgenme basamağına
sahip elementlerin veya farklı kimyasal bileşik halindeki elementler farklı toksik
özellik veya fayda göstermektedir. Bu nedenle elementin toplam miktarından daha
çok türlerin miktarlarının ayrı ayrı hesaplanması gerekli hale gelmiştir.
Eser element türleri örnek ortamında farklı karaklılık gösterirler ve daha karalı
yapıda olma eğilimindedirler. Türlerin miktarları ortamın sıcaklığı, pH’sı gibi
değişkenlerden etkilenmektedir.
Türleme çalışmalarında tayinden önce türlerin birbirinden ayrılabileceği koşulların
belirlenmesi gerekir. Genellikle iki şekilde uygulama vardır. Birinci uygulamada,
farklı türler için farklı deney koşullarında ayırma sağlanabiliyorsa, bu koşullar
uygulanarak farklı türler birbirinden ayrılır ve türler uygun bir tayin yöntemi ile ayrı
10
ayrı tayin edilir. İkinci uygulamada, eğer sadece türlerden birinin ayrılması ve tayini
için uygun koşullar belirlenebiliyorsa, önce ayırma işlemi uygulanmadan türlerin
toplam miktarı tayin edilir. Sonra belirlenen deneysel koşullarda örnekten ayrılan tür
tayin edilir. Daha sonra toplam miktardan tayin edilen türün miktarı çıkarılarak tayin
edilmeyen türün miktarı hesaplama yoluyla bulunur [Erdoğan, 2011]. Farklı türlerin
farklı deneysel koşullarda sinyal verdiği elektroanalitik teknikler gibi tekniklerde ise
her tür aynı çözeltide ayırma yapılmaksızın tayin edilebilir.
2.2. Katı Faz Özütleme Yöntemi ile İlgili Bazı Çalışmalar
2.2.1. Katı faz özütleme yöntemiyle yapılan zenginleştirme çalışmaları
Zenginleştirme
basamağının
esas
amacı,
gözlenebilme
sınırını
düşürmek,
girişimlerden kurtulmak ve tayin edilecek türün derişimini artırmaktır. Bazı
durumlarda bu amaçlar birbirleri ile çelişebilirler. Örneğin, çöktürme ile
zenginleştirme yönteminde çöktürücü reaktifin fazlası kullanılır. Çöktürücü reaktifin
fazlası geri kazanılan türün miktarını artırırken, tanık sinyalinin büyümesinden
dolayı gözlenebilme sınırının artmasına neden olabilir. Çöktürme ile zenginleştirme
yöntemlerinde bu iki faktör arasında en uygun koşulun belirlenmesi gereklidir. En
uygun koşulların belirlenmesi, zenginleştirme yönteminin ve tayin yönteminin tipine
bağlıdır [Alfassi ve Wai, 1992].
Katı faz üzerinde eser elementlerin tutunmasında fiziksel ve kimyasal adsorpsiyon,
iyon değiştirme ve kompleks oluşumu etkili olabilir. Bu mekanizmalar katı fazın
karakterine ve eser elementin kimyasal yapısına bağlıdır. Bu anlamda katı faz
özütleme yöntemi genelde iyon değiştirme ve adsorpsiyon olaylarına dayanır.
Zenginleştirme ve ayırma işlemlerinde katyon ve anyon değiştirici reçineler
kullanıldığı gibi şelat yapıcı iyon değiştiriciler de kullanılmaktadır. Bunlar arasında
Chelex–100, C–18, oktadesil bağlı silika-jel, selülozik iyon değiştiriciler sayılabilir
[Kendüzler, 2003; Janoš ve ark., 1992; Pozebon ve ark., 1998].
11
Tunçeli, katı faz olarak Amberlite XAD–16 kullanarak gümüş elementini
zenginleştirmiş ve alevli atomik absorpsiyon spektroskopisi ile tayini için yöntem
geliştirmiştir. Bu çalışmada, geri kazanma verimi %99,20, zenginleştirme faktörü 75,
gözlenebilme sınırı 0,047 mg/L ve adsorpsiyon kapasitesi de 4,66 mg/g olarak
bulunmuştur [Tunçeli ve Türker, 2000]. Tunçeli bir başka çalışmasında da yine
Amberlite XAD–16 kullanmış ve bu kez suda Cr(III) ve Cr(VI) türlemesini,
Cr(VI)’nın 1,5-difenilkarbazon kompleksini zenginleştirerek gerçekleştirmiştir.
Çalışmada, Cr(VI) için geri kazanma verimi %99,7, zenginleştirme faktörü 25,
gözlenebilme sınırı 45
g/L ve kullanılan reçinenin adsorpsiyon kapasitesi de 0,4
mg/g olarak bulunmuştur [Tunçeli ve Türker, 2002].
Çekiç ve ark., katı-faz özütleme yönteminde katı faz olarak o-aminobenzoik asit
immobilize edilmiş Amberlite XAD-4 reçinesinin kullanılabilirliğini incelemişlerdir.
Bu sentezlenen yeni reçinenin zayıf asidik ya da nötr sulu çözeltilerde eser
düzeylerde bulunan Pb(II), Cd(II), Ni(II), Co(II) ve
Zn(II)
metallerinin
zenginleştirilmesinde ve FAAS ile tayinlerinde uygulanabileceğini bulmuşlardır.
Adsorban üzerinde tutunan metalleri 1 mol/L HNO3 kullanarak geri almışlardır.
Yöntem, sentetik metal karışımlarının analizlerine, sertifikalı kömür ve göl suyu
örneklerine uygulanmıştır [Çekiç ve ark., 2004].
Tüzen ve ark., Dowex M 4195 kullanarak Au(III)’ün katı faz özütlemesini
gerçekleştirmiştir. Kullanılan şelatlaştırıcı reçinenin 100’den fazla çevrime dayanıklı
olduğu gözlenmiştir. Geri kazanma verimi % 95’den büyük olup, yöntemin
gözlenebilme sınırı 1,61
g/L, zenginleştirme faktörü ise 31 olarak bulunmuştur
[Tüzen ve ark., 2008].
Chen ve ark., çevre örneklerindeki V, Cu, Pb, Cd ve Hg elementlerinin on-line
yöntemle zenginleştirilmesinde kitosan modifiyeli mezoporus silikayı katı faz olarak
kullanmıştır. Elementler ICP-OES ile tayin edilmiştir. Geliştirilen yöntemde geri
kazanma verimi, tüm elementler için %90’dan fazla, zenginleştirme faktörü 20;
gözlenebilme sınırları sırasıyla 0,33 g/L, 0,30 g/L, 0,96 g/L, 0,05 g/L ve 0,93
12
g/L; adsorpsiyon kapasitesi sırasıyla 16, 3 mg/g, 21,7 mg/g, 22,9 mg/g, 12,2 mg/g
ve 13,5 mg/g’dır [Chen ve ark., 2009].
Sharma ve Pant, Amberlite XAD-16’yı gallik asit ile modifiye ederek şelatlaştırıcı
adsorban elde etmişlerdir. Hazırlanan adsorban Cr(III), Mn(II), Fe(III), Co(II), Ni(II)
ve Cu(II)’nin zenginleştirilmesi ve FAAS ile tayininde kullanılmıştır. Geliştirilen
yöntemde, geri kazanma verimleri tüm elementler için %95’ten fazla, zenginleştirme
faktörleri sırasıyla 300, 200, 400, 285,7, 300 ve 400, adsorpsiyon kapasitesi sırasıyla
216 mol/g, 180 mol/g, 403 mol/g, 281 mol/g, 250 mol/g ve 344 mol/g’dır
[Sharma ve Pant, 2009].
Ünsal ve Soylak, anot çamuru ve maden örneklerine eser düzeydeki altının tayini için
katı faz özütlemesine dayanan bir zenginleştirme yöntemi geliştirmiş ve FAAS ile
tayin etmiştir. Au(III) iyonları çift duvarlı karbon naotüp dolgulu kolon kullanılarak
örneklerden ayrılmış ve zenginleştirilmiştir. Metal iyonları 0,1 mol/L HCl ortamında
kolonda çift duvarlı karbon nanotüp üzerinde tutunduktan sonra asetonda 1 mol/L
HNO3 ile geri alınarak geri kazanım sağlamıştır. FAAS ile tayinde Au(III) için
gözlenebilme sınırını 1,5 µg/L olarak bulunmuştur [Ünsal ve Soylak 2011].
Soylak ve ark., tek duvarlı karbon nano tüpü katı faz olarak kullanarak Fe(III) ve
Cr(III) iyonlarını zenginleştirmiş ve FAAS ile tayin etmiştir. Geri kazanma veriminin
pH 8’de ≥ % 95 olduğu yöntemde Fe(III) ve Cr(III)’ün gözlenebilme sınırları
sırasıyla 2,12 ve 4,08 µg/L’dir [Soylak ve ark., 2010].
Soylak ve Ünsal, Fe(III) ve Pb(II) iyonlarının katı faz ekstraksiyonu ile ayrılması ve
zenginleştirilmesi için buki tüpler üzerine bir disk (BTs) oluşturmuşlardır. Fe(III) ve
Pb(II) iyonları pH 6’da nicel olarak geri almışlardır. Analitik değişkenlerin (örnek
hacmi, akış hızı gibi) etkilerini incelemişlerdir. Fe(III) ve Pb(II) için tayin sınırları
sırasıyla 1,6 ve 4,9 μg/L olarak bulmuşlardır. Yöntemin doğruluğunu belgeli referans
madde TMDA-51.3 ve katkılı su analizleri ile kontrol etmişlerdir. Fe(III) ve Pb(II)
13
için geliştirilen bu yöntemi doğal sulara ve bitkisel örneklere uygulamışlardır
[Soylak ve Ünsal, 2011].
Yalçınkaya ve ark., katı faz olarak sentezledikleri nano malzemeyi (ZrO2/B2O3)
kullanarak Co(II), Cu(II), Cd(II) iyonlarının zenginleştirilmesi ve FAAS yöntemi ile
tayini çalışmasını yapmıştır. Şelat oluşturucu maddelere ihtiyacın duyulmadığı
yöntemde, Co(II), Cu(II), Cd(II) iyonlarının gözlenebilme sınırları sırası ile 3,8, 3,3,
ve 3,1 µg/L, geri kazanma verimleri sırasıyla % 95 güven seviyesinde; % 96, % 95,
% 98 olarak verilmiştir. Kullanılan nano malzemenin adsorpsiyon kapasitesi ise Co
için 32,2 mg/g, Cu için 46,5 mg/g ve Cd için 109,9 mg/g’ dır [Yalçınkaya ve ark,
2010].
2.2.2. Literatürde yeralan bazı cıva türlemesi çalışmaları
Cıva (Hg) insan sağlığına ve ekosisteme etki eden en önemli toksik eser
elementlerden biridir. Cıvanın en büyük kaynağı insan kökenli atıklardır. 2000’den
fazla ticari ve sanayi araçta cıva kullanımına rastlarız. Cıva, piller, deterjanlar,
lambalar, tıbbi ürünler, böcek ilaçları (eskiden kullanılanlar), termometreler ve
termostat sondaları başta olmak üzere birçok malzemenin üretiminde veya üretim
sürecinde bulunmaktadır [Leopold ve ark., 2010; Çıtak, 2010].
Nüfus artışı ve kentleşme sonucu olarak daha fazla insan faaliyetleri önemli ölçüde
civa emisyonuna katkıda bulunur. Şu anda cıva kirliliği dünyada bir çevre sorunu
olarak kabul edilmektedir. Cıvanın toksitesi sadece toplam deişimine değil önemli
ölçüde kimyasal yapısına da bağlıdır [Ying Gao ve ark., 2012].
1953-1960 yılları arasında Japonya’da büyük bir suni gübre fabrikasının atıklarının
Minimata körfezine bırakılmasından dolayı bölgenin sakinlerinde bir takım hastalık
belirtileri görülmüştür. Konuşma zorluğu, uzuvlarda uyuşukluk, görme ve işitmede
bozukluklar gibi belirtilerle tahribata yol açan ve bu safhadan sonra da tedavi
edilemeyen bu hastalığa körfezin adına atfen “Minimata hastalığı” adı verilmiştir.
Birçok kişinin ölümüne ve sakat kalmasına yol açan bu hastalığın sebebinin körfeze
14
bırakılan cıvalı bileşikler ve bunlarla kirlenen balıkların yenmesi olduğu
anlaşıldıktan sonra, körfezde balık tutulması yasaklanmıştır. Benzer olaylar başka
ülkelerdede meydana gelmiştir. 1972 yılında Irak’ta cıvalı bileşiklerle kirlenmiş
buğday ve unların yenmesi sonucu 6530 kişi sehirlenmiş ve bunlardan 500 kadarı
hayatını kaybetmiştir [Leopold ve ark,. 2010; Çıtak, 2010].
Cıva doğada fiziksel veya kimyasal olarak birçok farklı yapıda bulunur. Elementel
Hg, inorganik Hg(II), metil cıva (CH3Hg+) ve dimetil cıva (CH3HgCH3) cıvanın en
önemli kimyasal yapılarıdır. Bunlardan en toksik olanı metil cıvadır. Humik
maddeler cıvayı metilleyebilir, Hg(II) iyonunu metalik cıvaya (Hg) indirgeyebilir ve
cıva ile kompleks bileşikler yapabilir. Cıva sedimentlerde metilleyici bakteriler etkisi
ile metil veya dimetil cıvaya dönüşebilir. Metil cıva bileşikleri de inorganik cıvaya
dönüşebilir [Leermakers ve ark., 2005; Çıtak, 2010].
Monometil cıva elementel cıva ve inorganik tuzlarına göre çok daha toksiktir.
Monometil cıva sindirim sistemi ile çok hızlı bir şekilde emilir önce kana sonrada
beyine ulaşır. Beyine ulaşan cıva sinir sisteminde geri dönüşü olmayan hasara yol
açar. Ayrıca civa bileşikleri proteinde bulunan –SH grubundaki kükürt atomlarına
kuvvetli kovalent bağlarla bağlanır. Bu bağlamda alkil-cıva bileşikleri, -SH
grubundaki hidrojen (H) atomlarıyla yer değiştirir. Cıva atomunun bu şekilde
proteine bağlanmasıyla, proteinin özelliklerinde ciddi bir değişme meydana gelir.
Bilindiği gibi proteinler hücre içinde enzim olarak hareket ederek organizmadaki
biyolojik tepkimeleri katalizlemede ve hücre zarının oluşumunda önemli bir rol
oynarlar.
Proteindeki
bileşiklerinden
cıvanın
kükürt
(S)
bağlanması
atomlarına
sonucunda
ortamda
proteinin
bulunabilecek
enzim
cıva
aktivitesi
durdurulmuş olur. Böylelikle hücre zarındaki madde geçişi engellenmiş olup hücre
zarı görevini yapamaz hale gelir [Leemakers ve ark., 2005; Çıtak, 2010].
Sanayi ürünleri, gıda ve su kirliliği kontrolünde toplam cıva analizinin µg/L
düzeyinde yapılması gerekmektedir. Cıva türlerinin ise toksisite ve bünyelerde
davranışları farklı olduğu için µg/L veya daha düşük derişimlerde analizleri
15
istenmektedir. Bu eser düzeydeki toplam cıva analizleri için en yaygın kullanılan
teknikler soğuk civa buharı oluşturma sonrası AAS, AFS, ICP-OES ve ICP-MS’ tir.
Cıvanın yukarıda bahsedilen özelliklerinden dolayı çevre, gıda vb. çeşitli örneklerde
cıva tayini gittikçe önem kazanmaktadır.
Krishna ve ark., inorganik ve metil cıva türlerini zenginleştirmek için polianilin
dolgulu kolon kullanmışlardır. MeHg iyonları % 0,3’lük HCl ile Hg(II) iyonları ise %
0,3’lük HCl içinde % 0,02’lik tiyoüre ile geri alınmıştır. Türlerin zenginleştirme
faktörü 120 ve 60 olarak bulunmuştur. Geliştirilen yöntemi doğal su örneklerine
uygulamıştır [Krishna ve ark., 2005].
Torres ve ark., yaptıkları bir çalışmada biyolojik örneklerde toplam cıva ve inorganik
cıva tayini için CVAAS yöntemini geliştirmişlerdir. İnorganik civa miktarını bulmak
için örnekler TMAH (tetrametilamonyum hidroksit) ile muamele edilerek çözülmüş,
toplam cıva miktarını bulmak için ise örnekler asit çözeltisiyle mikrodalga fırında
çözülmüştür. Toplam cıva ve inorganik cıvanın arasındaki farktan metil cıva
miktarını hesaplamışlardır. Yöntem belgeli referans madde (CRM), köpekbalığı
karaciğeri ve midye dokusuna başarıyla uygulanmıştır. Gözlenebilme sınırı inorganik
cıva için 0,02 mg/g, toplam cıva için ise 0,04 mg/g olarak bulunmuştur [Torres ve
ark.,2009].
Tüzen ve ark., Dowex Optipore SD-2 reçinesine streptococcus pyogens yüklenmiş
bir katı faz oluşturmuşlar, bu katı faz üzerinde inorganik ve organik cıva türlemesi ve
zenginleştirmesi yapmışlardır. pH 8’de sırasıyla MeHg iyonlarını 0,1 mol/L HCl ile
Hg(II) iyonlarını ise 2 mol/L HCl çözeltisiyle geri almışlardır. Cıva miktarının
belirlenmesi için CVAAS yöntemini kullanmışlardır. Bazı alkali, toprak alkali ve
geçiş metallerinin girişim etkileri de incelenmişlerdir. Hg(II) ve MeHg için
gözlenebilme sınırı sırasıyla 2,1 ng/L ve 1,5 ng/L olarak bulunmuştur. Yöntemde her
iki analit için zenginleştirme faktörünü 25 olarak bulmuşlardır. Yöntemin
doğruluğunu araştırmak için belgeli referans madde (NRRCCC-DORM 2 Dogfish
16
Muscle) analizini yapılmışlardır. Araştırmacılar yöntemi doğal su ve çevre
örneklerine uygulamışlardır [Tüzen ve ark., 2009].
Mousavi ve ark., cıva zenginleştirmesi ve tayini için, akışa enjeksiyon (FI)
yöntemiyle birleştirilen soğuk buhar atomik absorpsiyon spektrometresini (CVAAS)
kullanmışlardır.
Katı
bir
faz
üstünde
tutunan
cıva
(ng/L)
düzeylerini
belirlenmişlerdir. Deney için uygun koşulları (örnek hacmi, akış hızı, yabancı
iyonların etkisi ve kolonun kullanma kapasitesi) belirlemişlerdir. Gözlenebilme
sınırını 3 ng/L, dinamik aralığı 50 mL örnek için 10-250 ng/L olarak belirlemişler.
Bu yöntemi su ve atık su örneklerinde cıva tayini için uygulamışlardır [Mousavi ve
ark., 2010].
Mladenova ve ark., yüzey sularında düşük derişimlerde (µg/L) çözünmüş inorganik
cıva ve metil cıvanın ayrılması ve zenginleştirilmesi için L-sistein aşılanmış silikajel
sorbenti kullanmışlardır. Cıva türlerinin tayini için sürekli akışlı kimyasal buhar
oluşturmalı atomik absorpsiyon spektrometresi (CF-CVG-AAS) ve indüktif eşleşmiş
plazma–kütle spektrometresi (ICP-MS) yöntemlerini kullanmışlardır. Gözlenebilme
sınırını CF-CVG-AAS yöntemiyle Hg(II) için 1,5 ng/L MeHg için ise 5 ng/L olarak,
ICP-MS yöntemiyle ise Hg(II) için 1 ng/L, MeHg için ise 2,5 ng/L olarak
bulmuşlardır [Mladenova ve ark., 2012].
Dressler ve ark., yaptıkları çalışmada kırmızı şarapta toplam cıva, inorganik cıva ve
metil cıva tayini yapmak için akış enjeksiyonlu soğuk buhar oluşturmalı indüktif
eşleşmiş
plazma–kütle
kromatografi–indüktif
yöntemlerini
spektrometri
eşleşmiş
kullanmışlardır.
(FI-CVG-ICP-MS)
plazma–kütle
Geri
kazanım
yöntemi
spektrometri
değerleri
ve
gaz
(GC-ICP-MS)
%99-104
arasında
değişmektedir. GC-ICP-MS yönteminde gözlenebilme sınırlarını Hg(II) ve MeHg
için sırasıyla 0,77 ve 0,80 μg/L olarak bulmuşlardır. Toplam cıva tayini için
kullanılan FI-CVG-ICP-MS yönteminde gözlenebilme sınırını 0,01 μg/L olarak
bulmuşlardır. Örneklerde toplam cıva tayini yapılmış ve en yüksek değer olarak 0,55
± 0,02 μg/L bulmuşlardır [Dressler ve ark., 2012].
17
Zhang ve Adeloju, biyolojik örneklerde cıva tayini için hızlı bir akışa enjeksiyon
sistemli katalitik soğuk buhar atomik absorpsiyon spektrometri (FI-CCV-AAS)
yöntemi geliştirmiş inorganik ve organik karışımdan toplam cıva tayini için NaBH4
ile cıva türlerini indirgeyerek cıva buharı oluşturmuştur. Balık örneklerinde metil
cıva ve inorganik cıva, tiyoüre varlığında HCl ile FI-CCV-AAS yöntemiyle nicel
olarak geri kazanılmıştır. Yöntem balık karaciğeri referans maddesine (DOLT-4)
uygulanmış ve toplam cıva için % 91,5 metil cıva için ise % 102,3 geri kazanım elde
edilmiştir. Akışa enjeksiyon sisteminin kullanımı ile saatte 180 örnek analiz
edilmiştir [Zhang ve Adeloju, 2012].
Sarıca ve Türker, yaptığı çalışmada inorganik cıva ve metil cıva tayini için tepe
boşluklu tek damla mikro özütleme (HS-SDME) ile birleştirilen elektrotermal atomik
absorpsiyon spektrometri (ETAAS) yöntemini geliştirmiştir. Tiyoüre ve amonyum
prolidinditiyokarbamatı (APDC) alıcı faz olarak tespit etmiştir. Yöntemin
değişkenleri; mikroözütleme süresi, sıcaklık, NaBH4 derişimi, alıcı faz derişimi ve
ortam pH’sı farklı cıva türleri için ayrı ayrı koşullar belirlemek için araştırılmıştır.
Olası girişim etkileride incelenmiştir. Yöntemin doğruluğu belgeli referans madde
analizi (BCR 453 Tuna fish) ve katkılı numune analizleri ile doğrulanmıştır. Önerilen
yöntem ile cıva türlerinin tayini ve türlemesi için su ve balık örneklerine
uygulanmıştır. Gerçek örneklerde civa türlerinin derişimi %10’dan küçük bağıl
standart sapma ile bulunmuştur [Sarıca ve Türker, 2012].
2.3. Atomik Absorpsiyon Spektroskopisi (AAS) Yöntemi
2.3.1. Genel bilgiler
Atomik absorpsiyon spektroskopisi, elektromanyetik ışımanın gaz fazındaki serbest
atomlar tarafından absorplamasına dayanır. Eo temel enerji seviyesindeki bir atom,
h
enerjili bir foton absorplarsa, Ei uyarılmış enerji seviyesine geçer. Bu olaya
atomun uyarılması denir ve geçişe ait enerji değeri Planck eşitliğiyle hesaplanır:
18
Ei – Eo = h
=h
c
(2.1)
Burada;
h
: Planck sabiti, 6,626.10-34 J s
c
: Işık hızı, m/s
: Absorplanan ışının frekansı, s-1
: Absorplanan ışının dalga boyu, m
dir.
Buna göre bir atomun absorpsiyon yapması için, temel ve uyarılmış seviyeler
arasındaki enerji farkına denk enerjiye sahip bir ışın ile karşılaşması gerekir. Diğer
taraftan Lambert, homojen bir ortamdan geçen ışın şiddetinin, ışınların geçtiği
yöndeki ortamın kalınlığıyla üstel şekilde azaldığını, ancak ortama gelen ve geçen
ışın şiddetlerinin oranının gelen ışınların şiddetinden bağımsız olduğunu bulmuştur.
I = Io e-k'd
(2.2)
Burada;
Io
: Ortama gelen ışının şiddeti,
I
: Ortamı terk eden ışının şiddeti,
d
: Ortamın kalınlığı (ışının ortamdan geçtiği yol),
k'
: Absorpsiyon katsayısı (dalga boyu ve ortama bağlı bir katsayı),
dır.
Absorpsiyon katsayısı ışını absorplayıcı türün derişimi ile orantılıdır.
k' = k" C
Bu, bağıntı eşitlik 2.2’de yerine konur, düzenlenir ve her iki tarafın logaritması
alınırsa maddelerin ışın absorpsiyonuyla ilgili Lambert tarafından ortaya atılan ve
Beer tarafından geliştirilen Lambert – Beer yasası eşitliği ortaya çıkar.
19
ln (Io / I) = k"d C
k = k"/ 2,303 ise
A = log (Io / I) = k d C
(2.3)
Burada;
A
: Absorbans,
C
: Absorpsiyon yapan türün derişimi,
k
: Absorpsiyon katsayısı (Derişim molarite olarak alındığında molar
absorpsiyon katsayısı adını alır ve k yerine ε kullanılır.)
dır.
Absorpsiyon, temel haldeki atomların enerji absorplayarak uyarılmış hale geçmeleri
esasına dayandığından, absorbans da esas olarak temel haldeki atomların sayısına
bağlı olarak değişir. Belirli bir sıcaklıkta gaz fazında bulunan uyarılmış atomların
temel düzeydeki atom sayısına oranı Boltzmann eşitliğiyle hesaplanır.
Nu = No
gu
e
go
E/kT
(2.4)
Burada;
Nu, No : Sırasıyla uyarılmış ve temel düzeydeki atomların sayısı,
k
: Boltzmann sabiti,
E
T
: Uyarılmış ve temel düzeyler arasındaki enerji farkı,
: Mutlak sıcaklık,
gu, go : Sırasıyla uyarılmış ve temel düzeylerin istatistik ağırlıkları
dır.
Boltzmann eşitliğine göre ortamın sıcaklığı artarsa, temel düzeydeki atom sayısı
azalır ve uyarılmış düzeydeki atom sayısı artar. Atomik absorpsiyon spektroskopisi
tekniğinde, temel düzeydeki atom sayısının fazla olması istendiğinden, atomlaşma
sıcaklığı belirlenirken, bu sıcaklığın çalışılan element atomlarını atomlaştırmasına
20
fakat uyarmamasına dikkat edilmelidir. Genellikle, 3000 K’den düşük sıcaklıklarda
uyarılmış düzeydeki atom sayısı, temel düzeydeki atom sayısı yanında ihmal
edilebilir değerlerdedir. Bu nedenle bu sıcaklıklarda temel düzeydeki atom sayısı
ortamdaki toplam atom sayısına eşit alınabilir [Erdoğan, 2011].
2.3.2. Atomik absorpsiyon spektrometreleri
Atomik absorpsiyon spektrometrelerinde, atomlaştırılan tayin elementi üzerine onun
absorplayacağı ışın gönderilerek, atomlaştırıcıya gelen ve geçen ışın şiddetlerinin
oranı ölçülür. Bu işlemler için kullanılan bütün atomik absorpsiyon spektrometreleri
temelde aynı bileşenlere sahiptir. Atomik absorpsiyonun ölçülmesinde kullanılan
spektrometreler, esas olarak aşağıdaki bileşenlerden meydana gelir (Şekil 2.2).
Atomik absorpsiyon cihazlarının en önemli kısımlarından biri absorplanacak ışınları
yayan ışın kaynaklarıdır. Bu kaynaktan atomlaştırıcı ortamına tek dalga boylu ışın
gönderilmesine rağmen, atomlaştırıcı ortamından ilâve ışınların yayılması nedeniyle
atomlaştırıcıdan yayılan ışınlar çok dalga boyludur. İdeal bir atomlaştırıcının
emisyon yapmaması gerekirse de, bunun sağlanması uygulamada pek mümkün
değildir.
Absorpsiyon ölçmelerinde atomlaştırıcı tarafından yayılan ışınların etkisini gidermek
için tek ışın demetli alternatif akımlı sistemler geliştirilmiştir. Bu sistemlerde; ışın
kaynağı ile atomlaştırıcı arasına konan bir ışın kesici, kaynaktan gelen ışınları
dedektöre kesikli olarak gönderirken, atomlaştırıcıda oluşan ışınlar dedektöre sürekli
ulaşır. Kesikli gelen ışınlar dedektörde bir alternatif akım oluşturur ve elektronik
devreler de sadece bu akımı yükseltir. Böylece, ışın kaynağından gelen ışın
şiddetinin yanında atomlaştırıcıdan kaynaklanan ışın şiddeti ihmal edilmektedir. Işın
kaynaklarından kesikli ışınların gelmesi, kaynağa kesikli akım uygulamakla da
sağlanabilir (Şekil 2.2a).
21
Şekil 2.2. Atomik absorpsiyon spektrometresinin şematik gösterilişi [Welz ve
Sperling, 1999] 1. Işın kaynağı, 2. Atomlaştırıcı, 3. Monokromator, 4.
Dedektör, 5. Ölçme sistemi. (a. Tek ışın yollu, alternatif akımlı, b. Çift ışın
yollu, alternatif akımlı cihazlar.)
Işın kaynağından gelen ışınların kesikli hale getirilmesi yerine, ışın yoluna
yerleştirilen aynalı ışık biçer yardımıyla ışınlar bir kere atomlaştırıcıdan ve bir kere
de atomlaştırıcının dışından geçirilmek suretiyle de dedektöre ulaştırılabilir. Bu
şekilde alternatif akımlı çift ışın yollu cihazlar yapılmaktadır. Her iki ışın şiddeti
birbirine eşit olduğunda dedektörde herhangi bir akım meydana gelmemektedir.
Absorpsiyon nedeniyle ışın şiddetlerinin oranı değiştiğinde dedektörde bir akım
üretilmekte ve bu akım yükseltilerek ölçülmektedir. Bu sistemde çift ışın demeti
kullanılması nedeniyle kararlılık tek ışın demetli sistemlere göre daha iyi, fakat ışın
demetinin ikiye ayrılması yüzünden yayılan ışın şiddetinin azalması nedeniyle de
analitik duyarlık daha düşüktür (Şekil 2.2b) [Yalçınkaya, 2010].
2.3.3. Işın kaynakları
Atomik absorpsiyon hatlarının çok dar (0,002–0,005 nm) olması ve her elementin
kendisine has elektronik geçiş enerjilerinin bulunması atomik absorpsiyon
spektrometrik yöntemi oldukça seçici yapmaktadır.
22
Atomik absorpsiyon çalışmalarında kullanılan ışın kaynaklarına ait emisyon
hatlarının hat genişliğinin atomik absorpsiyon hattının hat genişliğine eşit veya daha
dar olması istenir. Bu nedenle atomik absorpsiyon spektrometrik yöntemde
absorpsiyon piklerinden daha dar bant genişliği olan ışın kaynakları kullanılır.
Atomik absorpsiyon ölçmelerinde kullanılan ışın kaynakları aşağıda listelenmiştir;
1. Oyuk katot lambaları (OKL)
2. Elektrotsuz boşalım lambaları (EBL)
3. Buhar boşalım lambaları
4. Alev
5. Sürekli ışın kaynakları
Bu tez kapsamında en yaygın kullanılan oyuk katot lambaları, elektrotsuz boşalım
lambaları ile son yıllarda kullanılmaya başlanan sürekli ışın kaynakları hakkında
bilgi verilmiştir.
Oyuk katot lambaları (OKL)
Atomik absorpsiyon spektroskopisinde halen en yaygın kullanılan ışık kaynağı oyuk
katot lambasıdır (Şekil 2.3). Bu kaynak, 1-5 torr arasında düşük basınca sahip argon
gibi bir inert gaz ortamında kapatılmış bir cam boruda tungsten veya nikel bir anot ve
silindir şeklinde bir katot içeren kapalı bir cam tüp şeklindedir. Katot, analiz
elementinin metalinden veya o metalin uygun bir alaşımından yapılmıştır.
Elektrotlar arasında uygulanan 300 V kadar bir potansiyel, inert gazın iyonlaşmasını
ve inert gaz iyonları ile elektronların elektrotlara yönelişinden doğan 5-10 mA’lik bir
akımı oluşturur. Potansiyel yeterince büyük ise, inert gaz katyonları katoda yeterli bir
enerji ile çarparak metal atomlarının bazılarını yerinden sökerek bir atom bulutu
oluşturabilir; bu işleme sıçratma adı verilir. Sıçratılan metal atomlarından bazıları
yine iyon ve/veya elektronların çarpmasıyla uyarılmış hale geçer ve sonra temel hale
dönerken katot metaline özgü dalga boyunda ışın yayarlar. Lambanın sıcaklığı
23
genellikle atomlaştırıcının sıcaklığından daha düşük olduğundan, lambadaki hat
genişlemesi atomlaştırıcıdakinden daha düşüktür. Bunun sonucu olarak lambada
yayılan ışının hat genişliği, atomlaştırıcıdaki atomların adsorpsiyon hat genişliğinden
daha dardır. Buda atomik absorpsiyon spektroskopisi için istenen bir durumudur.
Lambada katot malzemesinden sıçratılan metal atomları sonunda ya tekrar katot
yüzeyine ya da lambanın iç çeperlerine dönerek buralarda toplanır [Skoog ve ark.,
1999].
OKL’de katot elementine ait ışınlar yayıldığı için her element için o elementten
yapılmış katodu olan OKL gereklidir. Bazı durumlarda katot malzemesi olarak
alaşımlar kullanıldığında aynı lamba ile birkaç elementin tayini yapılabilmektedir.
Her element (bazen element grupları) için ayrı bir lamba gerektirmesi OKL’nin en
büyük yetersizliğidir. Son yıllarda her element için tek lambanın kullanıldığı sürekli
ışık kaynaklı cihazlar geliştirilmiştir.
Şekil 2.3. Oyuk katot lambasının şematik gösterilişi [Welz ve Sperling, 1999]
24
Elektrotsuz boşalım lambaları (EBL)
Eletrotsuz boşalım lambası, atomik çizgi spektrumu için yararlı bir kaynak olup aynı
element için yapılmış oyuk katot lambalarına göre 10-100 kat fazla ışın şiddeti
sağlayabilir. Tipik bir lamba, birkaç torr basınçlı argon gibi bir inert gaz ortamında,
analiz elementini metal veya bir tuzu şeklinde içeren kapalı bir kuvars borudur. Bu
kaynak elektrot içermez; bunun yerine şiddetli radyo frekansı veya mikrodalga
ışınım alanı ile gerekli enerji sağlanır. Bu alanda inert gaz iyonlaşır; iyonlar alanın
yüksek frekanslı bileşeni ile hızlandırılırlar ve böylece spektrumu istenilen metalin
atomlarını uyaracak enerjiye ulaşırlar [Skoog ve ark., 1999]. Uyarılan metal atomları
da kendilerine özgü dalga boylarında ışın yayar.
Sürekli ışın kaynakları
Son yıllarda tüm elementler için sadece bir ışık kaynağının kullanıldığı sürekli
kaynaklı atomik absorpsiyon spektrometreleri kullanılmaktadır. Böylece oyuk katot
lambasında olduğu gibi her element veya element grubu için ayrı lamba kullanımı
ortadan kalkmış ve tüm ultraviole-görünür bölgede (UV-GB) ışın yayan ışık
kaynakları kullanılmıştır. Işık kaynağı olarak ksenon ark lambalar kullanılarak ve
yüksek ayırma güçlü monokromatörler yardımıyla her piksel için yaklaşık 2 pm
çözünürlük
sağlanmaktadır.
Sürekli
ışın
kaynaklı
atomik
absorpsiyon
spektrometresinde 2 ayrı monokromatör kullanılmaktadır. Bunlardan bir tanesi
istenilen dalga boyunda ışının atomlaştırıcıya ulaşması için ışık kaynağı ile
atomlaştırıcı arasına diğeri ise atomlaştırıcı ile detektör arasına konmaktadır. Bu ışık
kaynaklarının kullanıldığı cihazlara, yüksek çözünürlüklü sürekli ışık kaynaklı
atomik absorpsiyon spektrometresi (HR-CSAAS) denilmektedir. [Welz ve ark,
2005].
25
2.3.4. Atomlaştırıcılar
Atomik adsorsiyon spektroskopisinde atomlaştırıcılar, çözeltideki analiz elementinin
serbest atomlarının oluşmasını sağlar. Absorpsiyon şiddeti gaz fazındaki serbest
atom derişimiyle doğru orantılı olduğundan, AAS’de atomlaştırıcıların çok önemli
bir işlevi vardır. Atomlaştırıcılar, genellikle alevli ve alevsiz (elektrotermal) olmak
üzere ikiye ayrılır.
Alevli atomlaştırıcılar
Örnekteki analiz elementini atomlaştırmak için uygun alev başlıklarıyla, çeşitli
yanıcı
ve
yakıcı
gazların
yakılmasıyla
elde
edilen
alevin
kullanıldığı
atomlaştırıcılardır. Genellikle alevli atomik absorpsiyon spektrometrelerinde yakıcı
gaz olarak hava, oksijen ve diazot monoksit (N2O), yanıcı gaz olarak da hidrojen
(H2), asetilen (C2H2) ve propan gazı kullanılır. Bazı alev türlerinin özellikleri Çizelge
2.1’de verilmiştir [Haswell, 1991].
Çizelge 2.1. Bazı alev türlerinin özellikleri
Yanıcı Gaz
Yakıcı Gaz
Alev Sıcaklığı, ( oC)
Yanma Hızı, (cm/s)
Asetilen
Hava
2300
160
Hidrojen
Hava
2050
320
Propan
Hava
1930
45
Hidrojen
N2O
2650
390
Asetilen
N2O
2950
285
Asetilen
Oksijen
3180
1130
Alevli atomlaştırıcılarda ön-karıştırmalı ve ön-karıştırmasız olmak üzere iki tür alev
başlığı kullanılır.
Ön-karıştırmasız (türbülent akımlı) alev başlıkları, nebülizörle birlikte imal edilmiştir
(Şekil 2.4). Örnek çözeltisi kılcal bir borudan yanıcı ve yakıcı gazların sağladığı
venturi etkisi ile emilir. Genellikle örnek akış hızı dakikada 1–3 mL’dir. Bu
26
başlıkların en önemli avantajı aleve daha çok örnek çözeltisinin gitmesini ve böylece
daha temsili bir analizin yapılmasını sağlamasıdır. Ön-karıştırmalı alev başlıklarına
göre patlama ve geri saçılma ihtimali daha azdır. Alev yolunun kısa olması ve bekin
tıkanma ihtimalinin fazlalığı ise önemli dezavantajlarıdır. Gürültülü çalışan bu
atomlaştırıcılar emisyon ve floresans çalışmalarında tercih edilirken, ışık yolunun
kısa olmasından dolayı absorpsiyonda kullanılmazlar.
Şekil 2.4. Ön-karıştırmasız alev başlığının şematik görünüşü [Robinson, 1990]
Ön-karıştırmalı (laminer akımlı) alev başlıklarında (Şekil 2.5) örnek çözeltisi yakıcı
gazın yardımıyla kılcal borudan emilir [Ebdon, 1982]. Örnekten gelen aerosoller
muhtelif engellere çarparak sis haline getirilir ve yakıt gazları ile birlikte aleve
taşınır. Örneğin büyükçe bir kısmı aleve taşınamaz ve dışarı atılır. Ön-karıştırmalı
alev başlıklarının ışın yollarının daha uzun olması sebebiyle duyarlık ve
tekrarlanabilirlik daha iyidir. Örnek emiş hızının ve aleve taşınma oranının
düşüklüğü bu alev başlıklarının yetersizliğidir.
27
Yakıcı gaz yardımı ile emilen çözeltinin aleve ulaşma oranı ve çözücünün
buharlaşma hızı örnek ve standart çözeltilerinin yoğunluk, viskozite ve yüzey
gerilimi gibi fiziksel özellikleriyle atomlaştırıcıda kullanılan sisleştiricinin yapısına
bağlı olarak değişir.
Sis halinde alev ortamına ulaşan çözeltideki çözünmüş maddeler ısı etkisiyle çözücü
buharlaştıktan sonra, kurur ve katı tanecikler oluşur. Çözücünün buharlaşma hızı,
damlacıkların büyüklüğüne ve çözücünün türüne bağlıdır. Katı tanecikler alevin
sıcaklığına bağlı olarak çeşitli değişikliklere uğrar. Organik maddeler yanar,
inorganik maddeler ise birbirleriyle veya alevdeki gazlarla tepkimeye girer. Alev
ortamında oluşan analiz elementinin tuzları sıcaklığın etkisiyle serbest atomlarına
dönüşür. Bu serbest atomlar kaynaktan gelen ışınları absorplayarak uyarılırlar. Alev
ortamında meydana gelen kimyasal olaylar çok karmaşıktır. Çözelti halinden
absorpsiyona kadar alev ortamında meydana gelen olaylar Şekil 2.6’da basit olarak
gösterilmiştir [Kendüzler, 2003].
Şekil 2.5. Ön-karıştırmalı alev başlığının şematik görünüşü
28
Şekil 2.6. MX çözeltisinin alevde atomlaşması ve uyarılması için olasılıklar
Alevli AAS’de kesin ve doğru analizler yapabilmek için alev şartlarının doğru olarak
tespiti gerekir. Alev başlığının yükseklik ve genişliğe bağlı olarak alevin sıcaklığı ve
bileşimi değişir. Bu nedenle tayin elementinin serbest atomlarının ve oksitlerinin
oluşumuna ve çözeltideki anyonlara bağlı olarak absorpsiyonun ölçüleceği uygun
alev profili seçilir. Absorpsiyon ölçümü kolaylıkla serbest atomları oluşturulan ve
29
hemen yükseltgenen elementler (Mg gibi) için başlığın dip kısmında, serbest
atomların oluşumu ve yükseltgenmeleri geç olan elementler (Ag gibi) için alevin üst
kısmında ve diğer elementler için de bu özelliklerine bağlı olarak alevin uygun
bölgesinde yapılır.
Alevi oluşturan gazların oranının da absorpsiyon ölçmelerinde büyük bir önemi
vardır. Alevlerde genellikle yakıcı gaz stokiyometrik orandan fazla kullanılmakta ve
bu durumda ortamda fazla oksijen bulunmaktadır. Kararlı oksitlerinin oluşumu
nedeniyle atomlaşma verimi azalan elementlerin analizine ortamda oksijenin
kalmaması için, yakıcı gaz stokiyometrik orandan daha az kullanılmalı ve yakıtın
tamamı yanmayacağı için de gerekli önlemler alınmalıdır [Erdoğan, 2011].
Elektrotermal atomlaştırıcılar
Alevli atomlaştırıcıların ön-karıştırmalı alev başlıklarında örneğin küçük bir kısmının
aleve taşınması, ön-karıştırmasız alev başlıklarında da yeterli atomlaşmanın
olmaması ve atomların alev içindeki ışın yolunda kalma sürelerinin çok kısa (10-4 s)
olması bu atomlaştırıcıların önemli dezavantajlarıdır. Bu mahsurları gidermek için
1970’li yıllarda elektrotermal atomlaştırıcılar kullanılmaya başlanmıştır (Şekil 2.7).
Bu atomlaştırıcılarda atomlaşma süresinin çok kısa ve atomların ışın yolunda kalma
sürelerinin daha uzun olmasından dolayı duyarlık; alevli yöntemlere göre en az 1000
kat daha iyidir.
30
Şekil 2.7. Grafit fırınlı atomlaştırıcının şematik görünüşü [Skoog ve ark., 1999]
Günümüzde en çok kullanılan elektrotermal atomlaştırıcılar grafit fırınlardır. Bu
fırınlar, genellikle 5–10 mm çapında, 20–30 mm uzunluğunda ve 1 mm kalınlığında
iki ucu açık silindir görünümündedir.
Elektrotermal atomlaştırıcılarda atomlaşma bir kaç ön basamaktan sonra gerçekleşir.
Birkaç mikrolitre hacmindeki örnek elektrik akımıyla ısıtılan karbon, tantal veya
iletken bir yüzey üzerinde düşük sıcaklıkta kurutulur (çözücü buharlaştırılır), sonra
sıcaklık yükseltilerek örnek kül edilir. Küllendirmeden sonra mümkün olan en kısa
sürede akım artırılarak sıcaklık atomlaşma sıcaklığına yükseltilir ve tayin
elementinin serbest atomlarının oluşması sağlanır. Atomlaşma basamağı dışındaki
tüm ısıtma basamaklarında grafit tüpün iç ve dış kısmından inert bir gaz geçirilerek
tüpün yanması engellenir. Atomlaşma kademesinde inert gazın kesilmesinin sebebi;
oluşan serbest atomların ışın yolunda daha uzun süre kalmalarını sağlayarak
duyarlığı artırmaktır.
Grafit fırınlı atomlaştırıcıların, grafit fırının ısıtılması için ayrı bir güç kaynağına
ihtiyaç duyulduğundan daha pahalı olmasına rağmen, alevli atomlaştırıcılara göre
birçok üstünlükleri bulunmaktadır. Bunlar;
31
Temel
halde
serbest
atomların
ışık
yolunda
alıkonma
süresi
alevli
atomlaştırıcılardaki alıkonma süresinden fazla olduğu için, duyarlık alevli
atomlaştırıcılara
göre
atomlaştırıcılarla
102-105
μg/mL
kat
seviyesinde
daha
tayin
yüksektir.
Dolayısıyla
yapılabilirken
alevli
elektrotermal
atomlaştırıcılarla µg/L seviyesinde tayin yapılabilir.
Çok küçük örnek hacimleri tayin için yeterli olmaktadır. Genellikle bir ölçüm
için 5-50 L kullanılmaktadır.
Vakum UV bölgede spektrum veren elementlerin tayinleri için kısmen
uygundur.
Argon gazı vakum UV bölgede absorpsiyon yapmazken alev gazları yapar.
Yanıcı ve yakıcı kullanılmadığı için tayin elementi üzerine bu gazların bozucu
etkisi gözlenmez.
Grafit fırında elde edilen atomik buhar kimyasal ve ısısal olarak daha iyi kontrol
edilebilir.
Aleve püskürtülmesi güç olan viskozitesi yüksek sıvıların analizi mümkündür.
Toksik maddelerle çalışıldığında, çok az örnek kullanıldığından, daha az toksik
buhar oluşur.
Grafit fırınlı atomlaştırıcılarda, tekrarlanabilirliği düşük olmasına rağmen, katı
numunelerin de doğrudan analizi yapılabilir.
Otomatik örnek verme sistemi kullanılması ile sürekli ve kesintisiz analiz
yapılabilir.
Yanıcı gazlar kullanılmadığı için laboratuvar güvenliği bakımından uygundur.
Elektrotermal atomlaştırıcının bu üstünlüklerinin yanında aleve göre bazı
yetersizlikleri de vardır:
Analiz süresi daha uzundur. Tipik bir grafit fırın ısıtma programı kademeli
sıcaklık artışları sebebi ile yaklaşık iki dakika sürerken alevde analiz süresi 15
saniyedir.
Zemin absorpsiyonu yüksek ve girişim daha çoktur.
32
Kül etme basamağında sıcaklığın yüksekliği sebebiyle tayin elementi kaybı
olabilir.
Kesinlik zayıftır. Özellikle elle örnek verme durumunda tekrarlanabilirlik iyi
değildir.
Deneysel koşulların ayarlanması alevli atomlaştırıcılara göre daha zordur
Özellikle inert gaz ihtiyacı ve grafit tüp kullanım ömrünün sınırlı olması sebebi
ile pahalı bir yöntemdir.
Hidrür oluşturmalı atomlaştırıcılar
Hidrür sistemli atomlaştırıcılar, arsenik, kalay, selenyum, bizmut, germanyum ve
kurşun gibi hidrürleri oluşturulabilen elementler için kullanılır. Bu yöntemde tayini
yapılacak elementler bir indirgeyici ile (NaBH4, Zn) uçucu hidrürlerine dönüştürülür
ve aleve ya da ısıtılmış kuvars tüpe yollanır. Alevde girişimin çok olmasından dolayı
genellikle ısıtılmış kuvars tüp tercih edilir [Skoog ve ark.,1998]. Bu sistemde sabit
hızda akan numune çözeltisi yine sabit hızda akan tetrahidroborat ve taşıyıcı gaz ile
karıştırılır. Sıvı ve gaz ayrıldığında gaz fazındaki metal hidrür, hidrojen gazı ve
taşıyıcı gaz atomlaştırıcıya gönderilir. Sıvı ise atık kısmından yer çekimiyle veya
pompa yardımıyla atılır. Bu işlem bu elementler için gözlenebilme sınırını 10 ile 100
kat azaltır. Bu türlerin oldukça toksik olmaları sebebiyle, düşük derişimlerin tayini
oldukça önemlidir. Aleve ya da kuvars tüpe taşınma inert bir gaz (Ar, He) yardımıyla
yapılır. N2 kullanılmaz çünkü hidrür yerine nitrür oluşabilir. Ayrıca tayin
elementlerinin bazılarının absorpsiyon yaptığı dalga boyunda absorpsiyon yapabilir
[Berkkan, 2004].
Soğuk buhar atomlaştırıcılar
Herhangi bir ısıtma uygulanmaması dışında HGAAS yöntemine benzerlikler
gösteren bu yöntemle sadece civa tayini yapılmaktadır. Civanın oda sıcaklığında bile
buhar basıncının yüksek olmasından faydalanılan yöntemde öncelikle çözelti
içerisindeki cıva, Hg(II) haline dönüştürülür. Sonra çözeltiye indirgen Sn(II) veya
33
NaBH4 eklenerek Hg(II) metalik cıva haline indirgenir ve inert gaz olarak kullanılan
He veya Ar gazları ile atomlaştırıcıya taşınır. Atomlaştırıcı olarak HGAAS
tekniğinede olduğu gibi kuvars cam boru kullanılır. Cam borunun uzunluğu
sayesinde temel haldeki cıvanın alıkonma süresi yani cıvaya özgü dalga boyundaki
ışınla temas süresi artar ve yöntemin duyarlığı da artmış olur [Sarıca, 2011]. Soğuk
buhar atomik absorpsiyon spektrometresinin şematik gösterilişi Şekil 2.8’da
verilmiştir.
Şekil 2.8. CVAAS yönteminin şematik gösterilişi
2.3.5. Monokromatör
Oyuk
katot
lambalarından
tek
dalga
boylu
ışınlar
gelmesine
rağmen,
atomlaştırıcıdaki olası uyarılmalardan dolayı birden fazla dalga boyunda ışınlar
meydana gelmektedir. Bu nedenle atomlaştırıcıdan sonra tayin elementi hattının
diğer
hatlardan
ayrılması
gerekmektedir.
Bu
amaçla
AAS
cihazlarında
monokromatör adı verilen ve prizma veya optik ağ içeren düzenekler
kullanılmaktadır.
34
Atomik absorpsiyon spektrometrelerinde spektral hatların ayrılması sadece
monokromatöre bağlı olmayıp oyuk katot lambasının emisyon hattı genişliğine
(yaklaşık 0,002 nm) ve tayin elementinin rezonans absorpsiyon hattı genişliğine
(yaklaşık 0,005 nm) bağlıdır. AAS tekniğinde atomlaştırıcıda yayılan ışınlar yinede
çok fazla olmadığından spektrumu en karmaşık elementler için bile, 0,2 nm
ayırıcılığa sahip monokromatörlerin atomik absorpsiyon spektroskopisi için yeterli
olduğu görülmüştür.
2.3.6. Dedektör
Atomik absorpsiyon spektrometrelerinde ışık sinyalini elektrik sinyaline çevirmek
için fotoçoğaltıcı tüpler kullanılır. Foto çoğaltıcı tüpler, ışığa duyarlı bir katot
(fotokatot), oluşan elektronların sayısını artıran dinotlar ve anottan ibaret bir vakum
fotoselidir.
Monokromatörden gelen bir foton fotokatot yüzeyine çarparak bir elektron koparır
ve bu elektron uygulanan gerilim farkı ile birinci dinota doğru çekilir ve dinot
üzerine gerilimle orantılı bir kinetik enerji ile çarpar. Bu dinotdan çok sayıda ikincil
elektron fırlar. Bu ikincil elektronlar hızlandırılarak ikinci dinoda çarparlar ve çok
fazla sayıda elektron fırlatırlar. Bu işlem devam ederken sonuçta çok sayıda elektron
anoda ulaşır ve devreden okunabilecek seviyede akım geçer. Devreden geçen akım,
dedektöre düşen ışık şiddeti ile orantılıdır.
Fotoçoğaltıcıların kullanıldığı spektral bölge, katot üzerindeki ışığa duyarlı tabakanın
ve ışığı geçiren malzemenin cinsine bağlıdır. En çok kullanılan malzeme Cs-Sb türü
alaşımıdır. Yazıcı veya bilgisayarlar ile dedektörlerden alınan sinyal, tayin
elementinin absorbansı, derişim vb. şeklinde okunur.
2.4. Atomik Absorpsiyon Spektroskopisinde Girişimler
Bir örnekte tayin edilecek elementle birlikte başka elementler, moleküller ve
iyonların varlığı nedeniyle atomlaştırıcıda oluşan serbest atomların sayısının
35
değişmesi ve/veya hatların monokromatörün ayıramayacağı kadar yakın olması
halinde analiz sonuçları doğru değerden sapma gösterir. Analizi yapılan örneğin
fiziksel ve kimyasal özellikleri ile içinde bulunan türlerin absorpsiyon veya emisyon
hatlarından kaynaklanan tayin elementi sinyalinin değişmesine “girişim” denir
[Erdoğan, 2011]. Kaynağına göre girişimler; fiziksel, kimyasal, iyonlaşma, spektral
ve zemin girişimi olarak sınıflandırılır.
2.4.1. Fiziksel girişimler
Deneylerde kullanılan standart ve örnek çözeltilerinin viskozite, yüzey gerilimi ve
yoğunluk gibi fiziksel özelliklerinin farklı olması halinde; birim zamanda aleve
taşınan çözelti hacmi, sisleşme verimi ve alevin sıcaklığı örnek ve standart çözelti
için farklı olur. Bu farklılık, birim hacimde çözelti başına alevde elde edilen serbest
atomların sayısının farklı olmasına neden olur. Bu nedenle karşılaştırılabilir sonuçlar
alınamaz.
Fiziksel girişimler örnek ve standart çözeltilerin fiziksel özellikleri birbirine
benzetilerek azaltılabilir. Bu da örneğin seyreltilmesiyle veya standart çözeltilerin de
aynı ortamda hazırlanması ile sağlanabilir. Bunun yanında analiz elementi özütleme,
iyon değiştirme, çöktürme gibi yöntemlerle ayrılarak fiziksel özellikleri standart
çözeltilerinkine benzeyen bir ortama da alınabilir ve fiziksel girişimler azaltılabilir.
Fiziksel girişimlerden kurtulmanın en iyi yöntemlerinden birisi de standart ekleme
tekniğini kullanarak tayin yapmaktır.
2.4.2. Kimyasal girişimler
Kimyasal girişimler, atomlaştırıcının yüzeyindeki katı fazda veya elementin
atomlaşması esnasında gaz fazında absorbans değerini değiştiren çeşitli kimyasal
tepkimelerin oluşması ile ortaya çıkar. Tayin elementinin moleküler halde zor eriyen,
zor buharlaşan bileşik oluşturması veya oluşan element atomlarının ortamdaki diğer
atomlarla veya radikallerle tepkimeye girerek kolay buharlaşan bileşik oluşturması
sonucu kimyasal girişimler gözlenir.
36
Birçok kimyasal girişim atomlaşma sıcaklığını artırarak veya kimyasal çevreyi
değiştirerek giderilebilir. Böylece güç atomlaşan bileşenler parçalanabilir ve düşük
sıcaklıkta girişim oluşturan bileşenler yok edilebilir. Ancak atomlaşma sıcaklığının
artırılması iyonlaşma girişimini artırabileceğinden en uygun atomlaşma sıcaklığı
belirlenmelidir.
Kimyasal girişimleri gidermenin diğer yolları şöyledir:
1. Tayin elementinin özütleme, iyon değiştirme, adsorpsiyon gibi yöntemlerle
girişim yapan örnek ortamından ayrılması,
2. Örnek ve standart çözeltilerin ortamlarının (matrikslerinin) birbirine benzetilmesi
3. Girişim yapan iyonlarla daha kararlı bileşik oluşturan maddeler katılarak tayin
elementinin serbest kalmasının sağlanması,
4. Tayin elementi ile daha kararlı yapı oluşturarak onun atomlaşmasını engelleyen
türlerden korunmasını sağlayan kompleks oluşturucuların katılması,
5. Standart ekleme yönteminin uygulanması
Alevsiz atomlaştırıcılarda inert ve indirgen bir ortam bulunduğundan, alevin
özellikleri sebebiyle ortaya çıkan bazı kimyasal girişimler bunlarda görülmez.
2.4.3. İyonlaşma girişimi
Tayin edilecek elementler, atomlaştırıcının sıcaklığı ve iyonlaşma gerilimine bağlı
olarak iyonlaşabilirler. İyonlaşma, temel haldeki serbest atom sayısını değiştirir.
İyonlarla temel haldeki atomların spektral özellikleri aynı olmadığı için iyon
lambadan gelen ışınları absorbe edemez ve tayin elementinin sinyali beklenenden
küçük olur.
Yakıcı gaz olarak N2O veya oksijenin kullanıldığı yüksek sıcaklık alevlerinde önemli
oranlarda iyonlaşma meydana gelirken, hava-asetilen gibi daha düşük sıcaklıklarda
iyonlaşma daha azdır. Bu nedenle, iyonlaşmanın etkisi düşük sıcaklıklı alevler
kullanılarak azaltılabilir. Fakat bu yöntem bütün elementler için uygun değildir.
Çünkü soğuk alevde de kimyasal girişimler artabilir.
37
İyonlaşma etkisi, ortama analiz elementinden daha kolay iyonlaşabilen başka
elementler (K, Cs gibi) katılarak giderilebilir. Böylece, kolay iyonlaşan element
nedeniyle ortamın elektron yoğunluğu artacağından, tayin elementinin iyonlaşması
engellenir.
2.4.4. Spektral girişimler
Spektral girişimler, analiz elementi dışındaki elementlerin absorpsiyon veya emisyon
hatlarının, tayin elementinin hattı ile üst üste çakışması veya monokromatorün
ayıramayacağı kadar yakın olması halinde ortaya çıkar. Oyuk katot lambalarının
emisyon hatları çizgisel olduğundan hatların çakışması neticesinde bozucu etkinin
meydana gelmesi çok nadir bir olaydır. Bu etkiler genellikle hatların 0,01 nm’den
daha yakın olması durumunda ortaya çıkar. Genel olarak spektral girişim varsa, tayin
elementinin bir başka hattı seçilir. Örneğin 308,211 nm’de hattı olan vanadyum,
308,215 nm’de hattı olan alüminyum tayinini bozar. Bu durumda alüminyum
309,270 nm’de tayin edilerek bozucu etki giderilir.
2.4.5. Zemin girişimi
Atomlaştırıcı ortamında oluşan molekül ve radikallerin absorpsiyon ve emisyon
yapması ve küçük taneciklerin ışınları saçması nedeniyle tayin elementinin dalga
boyunda ölçülen absorbans değerlerinde artışlar meydana gelir. Zemin absorpsiyonu
olarak adlandırılan bu etkiler mutlaka düzeltilmelidir. Zemin absorpsiyonunu
düzeltmekte kullanılan bütün yöntemlerde toplam absorbans değeri ölçülür ve zemin
girişiminden kaynaklanan absorbans değeri, toplam absorbans değerinden çıkarılır.
Zemin girişimi aşağıda verilen zemin düzeltme yöntemleri kullanılarak giderilebilir.
- Dalga boyu değiştirme,
- Sürekli ışın kaynağı kullanma,
- Zeeman yöntemi,
- Smith-Hieftje yöntemidir.
38
Ticari aletlerde daha çok sürekli ışın kaynağı ve Zeeman yöntemi kullanılmaktadır.
Tez kapsamında bu iki zemin düzeltme yöntemi kullanıldığı için bu iki yöntem
hakkında kısaca bilgi verilecektir.
Sürekli ışın kaynağı yöntemi
AAS ile yapılan tayinlerde zemin düzeltme tekniği olarak en çok kullanılan yöntem,
oyuk katot lambası ile birlikte sürekli ışın yayan ışın kaynağı kullanımıdır. Sürekli
ışın kaynağı olarak hidrojen, döteryum ve ksenon lambalar kullanılmaktadır. Ancak
AAS’de en çok kullanılanı döteryum lambasıdır. Döteryum lambasından gelen
ışınlar ve oyuk katot lambasından gelen ışınlar, ışın yolu üzerindeki bir ışın dilici
sayesinde atomlaştırıcı ortamından sırası ile geçer. Oyuk katot lambasından gelen
ışınlar hem atomlar hemde moleküller tarafından adsorplanırken, döteryum
lambasından gelen ışın başlıca moleküller tarafından adsorplanır. Döteryum
lambasından gelen ışınların atomlar tarafından adsorplanma kesri ihmal edilecek
kadar azdır. Bu nedenle oyuk katot lambasından gelen ışınlarla ölçülen adsorbans
değerinden (toplam absorbans), döteryum lambasından gelen ışınlarla ölçülen
absorbans değeri (zemin absorbansı) çıkararılarak atomik absorbans değeri
hesaplanır. Böylelikle moleküller ve şaçılmalardan kaynaklı zemin absorpsiyon etkisi
düzelmiş olur.
Zeeman yöntemi
Temel
haldeki
atomlara
manyetik
alan
uygulandığında
atomların
enerji
seviyelerindeki her bir elektronik geçişte birçok absorpsiyon çizgisinin oluşumuna
yol açan bir yarılma oluşur. Oluşan yeni çizgiler birbirinden 0,01 nm kadar ayrılır.
Bu olaya Zeeman etkisi denir. Manyetik alanın etkisiyle hatlar π, σ+ ve σ bileşenlerine ayrılır ve bu ayrılma sonucunda π bileşeni orijinal dalga boyunda
manyetik alana paralel, σ bileşenleri ise merkez bileşeninin iki yanında eşit dalga
boyu aralıklarında manyetik alana dik düzlemde sıralanır. Manyetik alan ışık
kaynağına uygulanırsa ışık kaynağının emisyonu, manyetik alana dik ve paralel
olarak polarize edilir. Işık kaynağının yaydığı π ve σ bileşenleri dedektöre ulaşmadan
39
önce dönen bir polarizörden geçerek birbirine dik olan bileşenlerine dönüştürülür. Bu
bileşenler atomlaştırıcıdan geçerken π bileşeni analitin atomları ve zemin girişimine
neden olan türler tarafından absorplanır. σ bileşenleri ise sadece zemin girişimine
neden olan türler tarafından absorplanır. π ve σ bileşenlerinin neden olduğu
absorbansların farkı ölçülerek analite ait absorbans değeri hesaplanır [Yıldız ve
Genç, 1993].
2.5. AAS Yöntemi ile Nicel Tayin
AAS yöntemi ile metalik özellik gösteren yaklaşık 70 kadar elementin nicel tayinini
yapmak mümkündür. Serbest haldeki tüm element atomları, üzerlerine düşen
kendilerine özgü dalga boylarındaki ışınları absorplarlar. Spektroskopik tayinlerde
elementin bu absorpsiyon dalga boylarından biri kullanılır. Genellikle seçilen dalga
boyu, absorpsiyonunun en şiddetli olduğu dalga boyu olup, bu dalga boyu rezonans
dalga boyudur. Böylece seçilen dalga boyunda küçük derişimlerde bile absorbans
değerleri okunabilir. Tayin ortamında elementin rezonans hattıyla spektral girişim
oluşturan element veya moleküller varsa, girişimin olmadığı fakat absorpsiyon
şiddetinin derişim tayini için yeterli olabileceği başka bir absorpsiyon hattı seçilir.
AAS yönteminde bir elementin nicel tayini, derişimleri bilinen standart çözeltilerin
absorbanslarıyla örnek çözeltisinin absorbansı karşılaştırarak yapılır. Katı veya sıvı
örnekleri atomlaştırmadan önce, uygun çözeltileri hazırlanır. Örnek çözeltileri
hazırlanırken tayin elementinin atomlaşma verimini olumsuz yönde etkilemeyen,
girişimlerin olmadığı ve yeterli absorpsiyon şiddetinin alınabileceği ortam şartları
ayarlanmalıdır. Örnek ve standartların absorbansları, cihazın bütün parametreleri
ayarlandıktan sonra aynı şartlarda ara verilmeden ölçülmelidir. AAS’de örnek
çözeltisinin derişimini belirlemek amacıyla aşağıdaki iki farklı yöntem izlenir.
2.5.1. Kalibrasyon yöntemi
Lambert-Beer yasasına göre absorbans, kuramsal olarak derişimle doğru orantılı
olarak değişir. Bu yöntem için, tayin edilecek elementin standart çözeltilerinden
40
belirli derişimlerde en az üç kalibrasyon çözeltisi hazırlanır. Kalibrasyon ve örnek
çözeltilerinin absorbansları önceden belirlenen dalga boylarında ölçülür. Kalibrasyon
çözeltilerinin derişimlerine karşılık absorbansları grafiğe geçirilir ve elde edilen
noktalar birleştirilerek bir doğru çizilir. Bu grafiğe “kalibrasyon eğrisi” denir.
Kalibrasyon eğrisinin doğrusal olduğu aralıktan (doğrusal çalışma aralığı)
faydalanılarak, örneğin absorbans değerlerine karşı gelen derişim bulunur. Örnek
çözeltilerinin absorbansları bu aralığın dışında ise, seyreltme veya deriştirme yolu ile
bu aralığa çekilmelidir. Çok sayıda örnek çözeltisine uygulanabilmesi bu yöntemin
üstünlüğüdür.
2.5.2. Standart ekleme yöntemi
Genellikle tayini yapılacak örnek çözeltilerinin karmaşık analiz ortamına sahip
olması, kalibrasyon çözeltilerinin ise benzer ortama sahip olmaması veya
benzetilememesi kalibrasyon grafiği yöntemi ile tayinler için önemli bir sorundur.
Böyle çözeltilerin analizinde ortamdan kaynaklanabilecek olan girişimler nedeniyle
doğru sonuçlara ulaşmak oldukça güçtür. Böyle durumlarda standart ekleme yöntemi
kullanılarak daha doğru sonuçlara ulaşmak mümkündür. Standart ekleme yönteminde
tayini yapılacak örnekten en az üç eşit kısım alınır. Birinci kısma yalnızca çözücü,
diğerlerine ise artan miktarlarda tayin edilecek elementin standart çözeltilerinden
katılıp her biri çözücü ile eşit hacme tamamlanır. Çözeltilerin absorbansları okunur
ve katılan elementin derişime karşı absorbans grafiği çizilir. Elde edilen doğrunun
derişim eksenini kestiği noktanın absorbans eksenine olan uzaklığı örnekteki analit
derişimine karşılık gelir (Şekil 2.9). Bu yöntem, analiz edilecek örneklerin sayısının
fazla olması halinde, kalibrasyon çözeltilerinin hazırlanması için çok fazla zaman
gerektirmesi sebebiyle kolay değildir, ancak fiziksel ve bazı kimyasal girişimleri
önlediği için daha doğru sonuçlar verir.
41
Şekil 2.9. Standart ekleme kalibrasyon grafiği örneği
2.6. Analitik Başarım ile İlgili Terimler
IUPAC (International Union of Pure and Applied Chemistry) ve ISO’nun (The
International
Organization
for
Standardization)
önerilerine
göre
analitik
spektroskopik yöntemlerde kullanılan analitik başarımla ilgili bazı terimler ve
tanımları aşağıda kısaca açıklanmıştır.
2.6.1. Duyarlık
Duyarlık teriminin en yaygın doğru tanımı, kalibrasyon duyarlığı yani analit
derişimindeki birim değişime karşı gelen sinyal şiddeti değişimidir. O halde
kalibrasyon duyalığı, kalibrasyon grafiğinin eğimi demektir. Kalibrasyon grafiği
doğrusal ise, duyarlık derişimden bağımsız, sabit bir değerdir. Grafik doğrusal değil
ise, duyarlık değişkendir; tek bir değerle verilmez.
Kalibrasyon duyarlığı, hangi derişim farkının gözlenebileceğini söylemez. Çünkü
derişimde
hangi
ölçekteki
bir
değişmenin
gözlenebileceğini
vermek
için
sinyallerdeki zemin gürültüsünün de hesaba katılması gerekir. Bu yüzden, kimi
zaman, analitik duyarlık denilen ikinci bir terim kullanılır. Analitik duyarlık,
kalibrasyon grafiği eğiminin, sabit derişim için analitik sinyalin standart sapmasına
42
oranıdır. Çoğu zaman, analitik duyarlık derişime kuvvetle bağlıdır [Skoog ve ark.,
1999].
2.6.2. Doğruluk
Doğruluk, “gerçek” veya gerçek kabul edilen değere yakınlık olarak tanımlanır ve
hata ile belirlenir. Analitik işlemlerde çeşitli hataların olması nedeni ile gerçek
değere ulaşmak mümkün değildir. Tayin elementi için ölçümün doğruluğu, standart
referans maddeler veya bağımsız analitik yöntemler kullanılarak kontrol edilir.
2.6.3. Kesinlik
Kesinlik, sonucun tekrarlanabilirliğinin bir ölçüsüdür. Çalışma şartlarında, uygulanan
analitik işlemlerin tekrarlanması ile elde edilen sonuçların birbirine yakınlığı,
kesinliği belirler. Kesinliğin en yaygın kullanılan ölçüsü standart sapmadır ( ).
2.6.4. Gözlenebilme sınırı
Gözlenebilme sınırı (limit of detection, LOD), tayin elementini içermeyen tanık
örneklerden elde edilen absorbans değerlerinin standart sapmasının üç katına karşılık
gelen derişim olarak tanımlanır. Derişim, elde edilen sinyal büyüklüğünün bir ölçüsü
olduğundan gözlenebilme sınırı duyarlığa bağlıdır. Ayrıca, genellikle gürültü diye
tanımlanan, zemindeki değişmeler olarak isimlendirdiğimiz ikinci bir değişkene de
bağlıdır.
Gözlenebilme sınırı aşağıdaki formül ile hesaplanır.
SLOD= Xt
k
t
Burada;
X t : Tanık çözelti sinyalleri okumalarının ortalaması,
(2.5)
43
SLOD : Gözlenebilme sınırı sinyal büyüklüğü,
σt: Tanık çözelti sinyallerinin standart sapması,
Bu çalışmada sinyal olarak absorbans ölçüldüğünden,
ALOD= Xt
k
t
yazılır
(2.6)
Kalibrasyon denklemi;
A= mC+ n ise,
Burada;
A: Absorbans,
n: Kalibrasyon doğrusunun absorbans eksenini kestiği nokta,
m: Kalibrasyon doğrusunun eğimi,
dir.
A yerine ALOD değeri yazılırsa, derişim cinsinden LOD (CLOD) hesaplananır.
ALOD= mCLOD + n
CLOD =
ALOD - n
m
(2.7)
(2.8)
Eşitlik 2.7’deki ALOD değeri eşitlik 2.8’de yerine konursa
CLOD =
Xt k t n
ifadesi elde edilir.
m
n ≈ X t kabulü yapılır ve % 95 için k=3 alınırsa,
(2.9)
44
(n ≈ Xt : Analit derişiminin sıfır olduğu durumda karşılık gelen n değeri ile kuramsal
olarak analit içermediği varsayılan tanık çözeltinin absorbans değeri yaklaşık olarak
birbirine eşit kabul edilir).
CLOD =
3 t
elde edilir.
m
(2.10)
Zenginleştirme işlemlerinin en önemli amaçlarından biri de çalışılan analitin
gözlenebilme sınırını düşürmektir. Bu nedenle, zenginleştirme çalışmalarında tayin
elementi için zenginleştirme yapılmadan bulunan gözlenebilme sınırı, analiz elementi
için bulunan zenginleştirme katsayısına bölünerek zenginleştirme yapılmış yöntemin
gözlenebilme sınırı bulunur.
2.6.5. Tayin sınırı
Tayin sınırı (limit of quantitation, LOQ), kullanılan yöntemin başarımıyla ile ilgili
bir başka terim olup tayinin yapılabileceği alt sınırı gösterir. Doğal olarak
gözlenebilme sınırı yakınlarında tayin yapılamaz. LOQ hesaplanırken LOD
hesaplanmasında kullanılan eşitlikte k yerine 10 alınır. Kabul edilebilir doğruluk ve
kesinlikte tayinler için örnekteki analit derişimi en az tayin sınırı değeri kadar
olmalıdır. LOQ değeri doğrusal çalışma aralığının alt sınırıdır.
CLOQ =
10 t
olarak verilir.
m
(2.11)
45
3. DENEYSEL KISIM
3.1. Cihaz ve Malzemeler
3.1.1. Atomik absorpsiyon spektrometresi
Bu çalışmada Perkin Elmer AA700 model Atomik Absorpsiyon Spektrometresi
kullanılmıştır. Cıva (Hg) tayini için cihaza takılmış MHS 15 hidrür sistemi
kullanılmıştır. Işın kaynağı olarak Perkin Elmer marka Hg elektrotsuz boşalım
lambası kullanılmıştır. CVAAS yöntemiyle cıva tayini için aletsel değişkenler
Çizelge 3.1’de verilmiştir.
Çizelge 3.1. CVAAS yöntemiyle Hg tayininde aletsel değişkenler
Değişken
Değer
Çalışan dalga boyu, nm
253,7
Lamba akımı, mA
4
Zemin Düzeltmesi
Döteryum Lambası
Hidrür oluşturma tepkime süresi, s
10
3.1.2. pH metre
Bu çalışmada pH ölçmeleri için, WTW marka 720 model dijital pH metre kullanıldı.
3.1.3. Çalkalamalı su banyosu
Bu çalışmada kesikli sistem çalışmaları için Nüve marka ST 402 model çalkalamalı
su banyosu kullanıldı.
46
3.1.4. Cam kolonlar
Bu çalışmada, çözelti haznesi 250 mL ve iç çapı 1 mm olan 15 cm uzunluğunda cam
kolonlar kullanıldı.
3.2. Reaktifler, Çözeltiler ve Kimyasal Maddeler
Deneylerde kullanılan çözeltiler, analitik saflıkta metal tuzları ve ultra saf su (18,3
MΩ.cm) kullanılarak hazırlandı.
3.2.1. Amberlyst 36
Kolon dolgu maddesi olarak katyon değiştirici Amberlyst 36 reçinesi (Rohm Haas)
kullanılmıştır.
3.2.2. Cıva (II) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L’lik Hg(II) (Hg(NO3)2) stok çözeltisi (J.T.Baker)
kullanılmıştır. 1000, 100 ve 10 µg/L’lik standart çözeltiler bu çözeltinin
seyreltilmesiyle hazırlanmıştır.
3.2.3. Metil cıva stok çözeltisi, 1000 mg/L
0,12g CH3ClHg (Sigma-Aldrich) 50 mL metanolde çözülerek hacmi 100 mL’ye
seyreltildi. 1000, 100 ve 10 µg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle
hazırlanmıştır.
3.2.4. Kobalt (II) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L kobalt stok çözeltisi (Merck) kullanılmıştır. 100, 50, 25 ve
5 mg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle hazırlanmıştır.
47
3.2.5. Nikel (II) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L nikel stok çözeltisi (Merck) kullanılmıştır. 100, 50, 25 ve 5
mg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.6. Sodyum stok çözeltisi, 1000 mg/L
0,9243 g NaNO3 (Carlo Erba) su ile çözülerek 250 mL’ye seyreltildi. 500, 250, 100,
50, 25 ve 5 mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.7. Potasyum stok çözeltisi, 1000 mg/L
0,6465 g KNO3 (Merck) su ile çözülerek 250 mL’ye seyreltildi. 250, 100, 50, 25 ve 5
mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.8. Mangan (II) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L Mn (II) stok çözeltisi (Merck) kullanılmıştır. 100, 50, 25
ve 5 mg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.9. Magnezyum stok çözeltisi, 1000 mg/L
2,5338 g MgSO4.7H2O (Merck) su ile çözülerek 250 mL’ye seyreltildi. 100, 50, 25
ve 5 mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.10. Çinko stok çözeltisi, 1000 mg/L
0,8338 g Zn(CH3COOH)2.2H2O (Merck) su ile çözülerek 250 mL’ye seyreltildi. 100,
50, 25 ve 5 mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
48
3.2.11. Kalsiyum stok çözeltisi, 1000 mg/L
1,4754 g Ca(NO3)2.2H2O (J. T. Baker) su ile çözülerek 250 mL’ye seyreltildi. 100,
50, 25 ve 5 mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.12. Bakır (II) stok çözeltisi, 1000 mg/L
0,9820 g CuSO4.5H2O (Merck) su ile çözülerek 250 mL’ye seyreltildi. 250, 100, 50,
25 ve 5 mg/L’lik çözeltiler bu stok çözeltinin seyreltilmesiyle hazırlanmıştır.
3.2.13. Amonyak çözeltisi, 0,1 mol/L
Yoğunluğu 0,91 g/mL olan % 25’lik (m/m) amonyak çözeltisinden (Merck) 0,75 mL
alınarak su ile toplam hacim 100 mL’ye tamamlandı.
3.2.14. Hidroklorik asit çözeltisi, 3,0 mol/L
Yoğunluğu 1,19 g/mL olan % 37’lik (m/m) hidroklorik asit çözeltisinden (Merck)
124,4 mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.15. Hidroklorik asit çözeltisi, 2,0 mol/L
Yoğunluğu 1,19 g/mL olan % 37’lik (m/m) hidroklorik asit çözeltisinden (Merck)
82,9 mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.16. Hidroklorik asit çözeltisi, 1,0 mol/L
Yoğunluğu 1,19 g/mL olan % 37’lik (m/m) hidroklorik asit çözeltisinden (Merck)
41,5 mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
49
3.2.17. Hidroklorik asit çözeltisi, 0,5 mol/L
Yoğunluğu 1,19 g/mL olan % 37’lik (m/m) hidroklorik asit çözeltisinden (Merck)
20,7 mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.18. Hidroklorik asit çözeltisi, 0,1 mol/L
Yoğunluğu 1,19 g/mL olan % 37’lik (m/m) hidroklorik asit çözeltisinden (Merck)
4,1 mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.19. Nitrik asit çözeltisi, 3,0 mol/L
Yoğunluğu 1,40 g/mL olan % 65’lik (m/m) nitrik asit çözeltisinden (Merck) 103,4
mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.20. Nitrik asit çözeltisi, 2,0 mol/L
Yoğunluğu 1,40 g/mL olan % 65’lik (m/m) nitrik asit çözeltisinden (Merck) 68,9
mL alınarak su ile toplam hacim 500 mL’ye tamamlandı.
3.2.21. KMnO4 çözeltisi, 0,1 mol/L
0,395 g katı KMnO4 (Merck) tartıldı ve hacmi su ile 25 mL’ye tamamlandı.
3.2.22. NaBH4 çözeltisi, % 0,5 (m/V)’lik (%0,5 (m/V)’lik NaOH çözeltisinde)
0,5 g NaBH4 (J.T. Baker) ve 0,5 g NaOH (Merck) tartıldı bir miktar su ile çözüldü ve
100 mL’lik ölçülü balonda su ile işaret çizgisine kadar tamamlandı.
50
3.2.23. Tiyoüre (3 mol/L HCl çözeltisinde), 0,05 mol/L
0,95 g Tiyoüre ( Acros ) tatıldı az miktarda suda çözdükten sonra 62,6 mL derişik
HCl çözeltisi eklenerek hacmi 250 mL’ye tamamlandı.
3.2.24. Tiyoüre (3 mol/L HCl çözeltisinde), 0,1 mol/L
1,9 g Tiyoüre ( Acros ) tatıldı az miktarda suda çözdükten sonra 62,6 mL derişik HCl
çözeltisi eklenerek hacmi 250 mL’ye tamamlandı.
3.2.25. Tiyoüre (1 mol/L HCl çözeltisinde), 0,2 mol/L
3,8 g Tiyoüre ( Acros ) tatıldı az miktarda suda çözdükten sonra 20,4 mL derişik HCl
çözeltisi eklenerek hacmi 250 mL’ye tamamlandı.
3.2.26. Tiyoüre (2 mol/L HCl çözeltisinde), 0,2 mol/L
3,8 g Tiyoüre ( Acros ) tatıldı az miktarda suda çözdükten sonra 40,8 mL derişik HCl
çözeltisi eklenerek hacmi 250 mL’ye tamamlandı.
3.2.27. Tiyoüre (3 mol/L HCl çözeltisinde), 0,2 mol/L
3,8 g Tiyoüre ( Acros ) tatıldı az miktarda suda çözdükten sonra 62,6 mL derişik HCl
çözeltisi eklenerek hacmi 250 mL’ye tamamlandı.
3.2.28. Arsenik(III) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L As(III) stok çözeltisi (Merck) kullanılmıştır. 50, 25, 10 ve
5 µg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle hazırlanmıştır.
51
3.2.29. Selenyum(IV) stok çözeltisi, 1000 mg/L
Bu çalışmada, 1000 mg/L Se(IV) stok çözeltisi (Merck) kullanılmıştır. 50, 25, 10 ve
5 µg/L’lik standart çözeltiler bu çözeltinin seyreltilmesiyle hazırlanmıştır.
3.3. Örneklerin Hazırlanması
3.3.1. Model çözeltiler
Civanın zenginleştirilmesi için en uygun deneysel şartların belirlemesinde kullanılan
model çözeltiler, otomatik pipet yardımıyla 100 µg/L’lik Hg(II) veya MeHg
çözeltilerinden alınan 2 mL çözeltinin saf su ile 50 mL’ ye tamamlanmasıyla
hazırlanmıştır. Böylelikle 50 mL’de 200 ng Hg(II) ve MeHg içeren model çözeltiler
hazırlanmıştır.
3.3.2. Su örneklerinin hazırlanması
Çeşme suyu örnekleri Ankara ili şebeke suyundan alındı. Kullanılmadan önce mavi
bant (sık gözenekli) süzgeç kâğıdından süzüldü. Kaplıca su örnekleri Ankara
yakınlarında Ayaş, Beypazarı ve Kızılcahamam ilçelerinden alındı. Su örneklerinin
alındıkları andaki sıcaklık ve pH değerleri Çizelge 3.2’de verilmiştir. Su örneklerine,
bakteri oluşumunu önlemek ve kararlı halde kalmalarını sağlamak amacı ile hacimce
% 1 olacak şekilde nitrik asit eklenmiştir.
Çizelge 3.2. Su örneklerinin alındıkları andaki pH değerleri ve sıcaklıkları
Örnek
T (oC)
pH
Beypazarı Tahtalı – Dutluk kaplıca suyu
50
≈ 6-7
Ayaş içmece suyu
52
≈7
Ankara şebeke suyu
21
≈7
Kızılcahamam kaplıca suyu
75
≈7
52
3.3.3. Balık örneğinin hazırlanması
Balık örneğinde cıva tayini yaparken, Hirfanlı barajından tutulan sazan balığı
(cyprinus carpio) kullanıldı. Mikserde parçalanarak etüvde kurutulan balık
örneğinden 1 g tartılıp 5 mL 5 mol/L HCl çözeltisiyle 30 dakika ultrasonik banyoda
özütlendi ve cıva iyonlarının çözeltiye geçmesi sağlandı. Çözelti ve katı kısım
santrifüj edildikten sonra dökülerek ayrıldı [ Rio-Segade ve Bendicho, 1999]. Alınan
çözelti 50 mL’ye seyreltildi. Balık örneği alınmadan diğer işlemler aynen
tekrarlanarak bir tanık örnek çözeltisi de hazırlandı.
3.3.4. Belgeli referans maddelerin hazırlanması
Çalışmanın doğruluğunu araştırmak için ERM-CE464 (TUNA FISH) Belgeli
Referans Maddesi kullanıldı. 0,1 g referans madde tartılıp 5 mL 5 mol/L HCl
çözeltisiyle 30 dakika ultrasonik banyoda özütlendi ve cıva iyonlarının çözeltiye
geçmesi sağlandı. Çözelti ve katı kısım santrifüj edildikten sonra dökülerek ayrıldı
[Rio-Segade ve Bendicho, 1999].
Alınan çözelti 50 mL’ye seyreltildi. Belgeli
referans madde alınmaksızın diğer işlemler aynen tekrarlanarak bir tanık referans
çözeltisi de hazırlandı.
3.4. Katı Faz Özütlemesi (SPE) ile Genel Zenginleştirme Yöntemi
Civanın zenginleştirilmesi, türlemesi ve bilinen bir ortama alınması amacı ile model
çözeltiler hazırlandı ve kolon tekniği ile zenginleştirme/türleme için en uygun deney
koşulları (pH, çözelti akış hızı vb.) belirlendi. Belirlenen bu koşullarda geliştirilen
yöntemin doğruluğu, kesinliği,
çalışma aralığı gibi analitik değişkenleri de
belirlendi.
3.4.1. Kolonun hazırlanması
Katı faz olarak kullanılacak, Amberlyst 36 reçinesinden 300 mg tartılıp, 15 cm
uzunluğunda, 1 cm çaplı musluklu cam kolonlara yerleştirildi. Adsorbanın çözelti
53
akışından etkilenmemesi ve dağılmaması için üstüne ve altına cam pamuğu
yerleştirildi. Adsorban, kullanımdan önce, örnek çözeltinin pH’sına eşit pH’da sulu
asit veya baz çözeltisi ile muamele edilerek şartlandırıldı. Kullanımdan sonra,
adsorban önce geri alma çözeltisiyle sonrada seyreltik HCl (0,5 veya 1 mol/L) ve su
ile yıkandı ve bir sonraki kullanıma kadar suda bekletildi. Kullanılan kolonun
şematik görünüşü Şekil 3.1’de verilmiştir.
Şekil 3.1. Katı faz özütleme tekniğinde kullanılan kolonun şematik görüntüsü
3.4.2. Zenginleştirme, türleme ve tayin yöntemi
Eser miktardaki cıvanın zenginleştirilmesi ve türlemesi için kolon tekniği kullanıldı.
Geliştirilen yöntemle Hg(II) ve MeHg iyonlarının her ikisi de pH 4’de Amberlyst
36’da tutundu. Bu nedenle türleme farklı geri alma çözeltileriyle önce MeHg sonra
Hg(II) geri alınarak yapıldı. Geri alma çözeltisi olarak MeHg için 10 mL 0,1 mol/L
54
HCl çözeltisi ve Hg(II) için ise 3 mol/L HCl içinde 0,2 mol /L tiyoüre çözeltisi
kullanıldı. Geri alma çözeltilerinin cıva içeriği CVAAS yöntemiyle tayin edildi.
CVAAS ile tayin işleminde Hg(II)’nin metalik cıvaya indirgenmesi için % 0,5
NaOH içinde % 0,5 NaBH4 çözeltisi kullanıldı. MeHg ise önce % 0,1’lik KMnO4 ile
muamele edilerek Hg(II)’ye dönüştürüldü ve sonra yukarıdaki işlem aynen
uygulanarak tayin edildi.
Ayırma ve zenginleştirmenin başarımını kontrol etmek için geri kazanma verimi
hesaplandı. Örnek çözeltisindeki cıvanın tamamının geri alma çözeltisine geçtiği
varsayılarak geri alma çözeltisindeki cıva derişimi kuramsal olarak hesaplandı. Bu
değer ile geri alma çözeltisinde CVAAS ile bulunan değerden yararlanılarak geri
kazanma verimi aşağıdaki şekilde hesaplandı.
% Geri kazanma verimi = (CVAAS ile bulunan derişim / Kuramsal olarak
hesaplanan derişim) x100
3.4.3. Adsorpsiyon kapasitesi tayin yöntemi
Kullanılan adsorbanın adsorpsiyon kapasitesinin tayini için katı-sıvı faz arasında
denge oluşması gerekli olduğundan kolon sistemi yerine kesikli sistem kullanıldı. Bu
amaçla, 5, 10, 25, 50, 100 ve 200 µg/mL Hg(II) ve MeHg iyonu içeren ayrı 50
mL’lik örnek çözeltilerinin pH değeri deneysel olarak belirlenen en uygun değere
ayarlandıktan sonra 0,1 g Amberlyst 36 içeren erlenlere ilave edildi. Dengeye
ulaşmaları için çözeltiler iki saat süreyle 120 rpm’de oda sıcaklığında çalkalandı.
Ardından her bir çözeltiden 10’ar mL alınarak, uygun seyreltme işlemi yapılarak cıva
içerikleri CVAAS yöntemi ile tayin edildi. Hg(II) ve MeHg iyonlarının denge
derişimine karşı katının gramı başına adsorplanan metal iyonu miktarı grafiğe
geçirilerek Langmuir adsorpsiyon grafiği elde edildi. Langmuir eşitliğinin aşağıda
verilen doğrusallaştırılmış haline göre CE’ye karşı CE/QE grafikleri çizildi. Bu
grafiklerden, doğrunun eğim ve kesim noktasından yararlanılarak Qo ve b Langmuir
sabitleri hesaplandı. Doğrusallaştırılmış Langmuir izotermi için bağıntı aşağıda
verilmiştir:
55
C
CE
1
= E +
QE
Q0 b
Q0
Burada;
CE: Dengede çözeltideki metal iyonu derişimi (mg/L),
QE: Dengede adsorbanın gramı başına adsorbe ettiği metal miktarı (mg/g),
b : Langmuir sabiti (L/mg),
Qo: Kapasite parametresi (adsorpsiyon kapasitesi) (mg/g)
dır.
56
4. SONUÇLAR
4.1. Cıva Türlemesi ve Tayini
4.1.1. CVAAS yöntemi ile cıva türlemesi ve tayini için en uygun yöntem
değişkenler
CVAAS yöntemiyle Hg tayininde, cıva türleri metalik cıvaya indirgenerek tayin
edilir. Bu çalışmada cıva iyonlarının metalik cıvaya dönüştürmek için NaBH4
çözeltisi kullanıldı. Çalışmada CVAAS yöntemiyle cıva tayini için kullanılan aletsel
değişkenler. Çizelge 3.1 de verilmiştir. Çalışmanın sonraki aşamalarında verilen bu
değerler kullanılmıştır.
Hg2+ + 2NaBH4 + 6H2O → Hg0 + 2Na+ + 2H3BO3 + 7H2 [Sarıca, 2011]
pH’nın geri kazanma verimine etkisi
Metal iyonlarının adsorpsiyonuna ve türlemesine etki eden en önemli etkenlerden
birisi de pH’dır. Bundan dolayı, Hg(II) ve MeHg iyonlarının geri kazanımları üzerine
pH’nın etkisi her bir iyon için ayrı ayrı olarak pH 2-10 arasında incelendi. Öncelikle,
en uygun pH değerini veya pH aralığını belirleyebilmek için seyreltik HCl ve/veya
seyreltik NH3 çözeltileri kullanılarak kolon dolgu maddesi sırasıyla pH 2, 3, 4, 5, 6, 8
ve 10’a şartlandırıldı. Takiben, ayrı ayrı 50 mL 100 ng Hg(II) ve 50 mL 100 ng
MeHg içeren çözeltiler hazırlandı. Hazırlanan bu çözeltilerin pH değerleri de
sırasıyla pH 2, 3, 4, 5, 6, 8 ve 10’a ayarlandıktan sonra çözelti akış hızı 3 mL/min’e
ayarlanarak kolondan geçirildi. Adsorban üzerinde biriktirildiği düşünülen MeHg ve
Hg(II) iyonları 10 mL 0,1 mol/L HCl çözeltisi ile geri alındı. Geri alma
çözeltilerindeki cıva iyonları derişimi CVAAS yöntemi ile tayin edildi ve cıva türleri
için geri kazanma verimleri hesaplandı. Bu koşullarda pH’nın cıva türlerinin geri
kazanma verimine etkisi Şekil 4.1’de görülmektedir. Şekil 4.1’den görüleceği üzere
MeHg için geri alma çözeltisi olarak 0,1 mol/L HCl çözeltisi kullanıldığında pH 3- 6
aralığında % 90’dan büyük geri kazanma verimi elde edilirken, Hg(II) için pH 2-10
57
aralığında geri kazanma verimi %10’un altında elde edildi. Geri alma çözeltisinde
Hg(II)’nin yeteri kadar bulunamaması üzerine Hg(II) iyonunun adsorbanda tutunup
tutunmadığını araştırmak için kolon süzüntüsünde de cıva tayini yapıldı ve süzüntüde
de yeteri kadar cıva belirlenemedi. Bu sonuçlardan Hg(II)’nin adsorbanda tutunduğu
ancak geri alma çözeltisinin uygun olmayışından dolayı adsorbandan geri
alınamadığı anlaşıldı. Bunun üzerine farklı geri alma çözeltilerinin denenmesine
karar verildi.
Şekil 4.1. Geri alma çözeltisi olarak 0,1 mol/L HCl çözeltisi kullanıldığında pH’nın
geri kazanma verimine etkisi
Özetle pH 4’te her iki cıva türü de adsorbanda tutunmakta ancak 0,1 mol/L HCl ile
metil cıva nicel olarak geri alınabildiği halde Hg(II) nicel olarak geri
alınamamaktadır. Bu özellikten yararlanılarak türleme yapılabileceği düşünüldü ve
Hg(II) iyonunun da geri alınabileceği farklı geri alma çözeltisi araştırıldı.
58
Şekil 4.2. pH’ nın cıva türlerinin geri kazanma verimine etkisi
Geri alma çözeltisinin geri kazanma verimine etkisi
Geri alma çözeltisinin cinsi ve derişimi katı faz özütleme sisteminin analitik başarımı
açısından önemlidir. Bu amaçla öncelikle HNO3 ve HCl çözeltileri geri alma
çözeltisi olarak denendi. Kullanılacak geri alma çözeltisinin sonraki tayin
basamağına uygun olması önemli olduğundan öncelikle inorganik asitlerin etkisi
incelenmiştir. İnorganik asitlerle Hg(II) için yeterli geri kazanma verimi elde
edilemediğinden farklı derişimlerde asitli tiyoüre karışımları da denendi. Sonuçlar
Çizelge 4.1 ve 4.2’de verilmektedir. MeHg için 10 mL 0,1 mol/L HCl çözeltisi
yeterli iken Hg(II) için yeterli olmadığı görülmektedir. Hg(II) için 10 mL 3 mol/L
HCl içinde 0,2 mol/L tiyoüre çözeltisinin nicel geri kazanma için yeterli olduğu
belirlendi.
59
Çizelge 4.1. Geri alma çözeltisi cinsi ve derişiminin Hg(II)’ nin geri kazanma
verimine etkisi
Geri alma çözeltisi
Geri kazanma verimia (%)
10 mL 2mol/L HNO3
-
10 mL 2 mol/L HCl
20±4
10 mL 3 mol/L HCl
49±5
10 mL 0,05 mol/L Tiyoüre (3 mol/L HCl’de)
55±3
10 mL 0,1 mol/L Tiyoüre (3 mol/L HCl’de)
87±4
10 mL 0,2 mol/L Tiyoüre (1 mol/L HCl’de)
20±3
10 mL 0,2 mol/L Tiyoüre (2 mol/L HCl’de)
62±3
10 mL 0,2 mol/L Tiyoüre (3 mol/L HCl’de)
99±2
5 mL 0,2 mol/L Tiyoüre (3 mol/L HCl ‘de)
74±2
a
Üç ölçümün ortalaması ± standart sapma
Çizelge 4.2. Geri alma çözeltisi cinsi ve derişiminin MeHg ‘nın geri kazanma
verimine etkisi
Geri alma çözeltisi
Geri kazanma verimia (%)
10 mL 2 mol/L HNO3
45±3
10 mL 3 mol/L HCl
99±3
10 mL 2 mol/L HCl
98±2
10 mL 1 mol/L HCl
97±3
10 mL 0,5 mol/L HCl
97±3
10 mL 0,1 mol/L HCl
96±4
5 mL 0,1 mol/L HCl
51±3
a
Üç ölçümün ortalaması ± standart sapma
Bu geri alma çözeltileri ile elde edilen geri kazanma veriminin pH’ya bağlılığı Şekil
4.2’de görülmektedir. Şekil 4.2’ye göre her iki cıva türünün de pH=4’te adsorbanda
tutunduğu anlaşılmaktadır. Şekil 4.1 ve Şekil 4.2 birlikte değerlendirildiğinde pH
4’te tutunan iki cıva türünün farklı geri alma çözeltileri kullanılarak kolondan geri
alınabilecekleri anlaşılmaktadır. Bu çalışmada bu özellikten yararlanılarak türleme
60
yapılmıştır. Aynı çözeltide aynı pH’da adsorbanda tutunan cıva türleri farklı geri
alma çözeltileriyle geri alınmış ve ayrı ayrı tayin edilmiştir. Geri alma çözeltisi için
sonraki çalışmalarda bu çözeltiler kullanılmıştır.
Örnek akış hızının geri kazanma verimine etkisi
Örneğin akış hızının cıva türlerinin geri kazanma verimine etkisi en uygun deney
koşulları altında (pH, geri alma çözeltisi gibi) incelendi. Cıva iyonlarının model
çözeltileri kolondan 1-4 mL/min akış hızlarında geçirildi ve akış hızına bağlı olarak
geri kazanma verimleri belirlendi. Şekil 4.3’de görüldüğü gibi geri kazanma verimi
her iki cıva türü için akış hızı 3 mL/min’den sonra düşmektedir. Bu nedenle cıva
iyonlarının ayrılması ve zenginleştirilmesi için en uygun akış hızı 3 mL/min olarak
belirlenmiştir.
Şekil 4.3. Akış hızının cıva türlerinin geri kazanma verimine etkisi
Örnek hacminin (veya analit derişiminin) geri kazanma verimine etkisi
En yüksek çözelti örnek hacminin belirlenmesi, yöntemle kaç kat zenginleştirme
yapılabileceğini görmek bakımından oldukça önemlidir. Bu amaçla cıva türlerinin
miktarı sabit tutularak örnek hacmi artırıldı ve bir seri örnek çözeltileri hazırlandı.
61
Her birinde 0,1 μg Hg(II) ve MeHg içeren 25, 50, 100, 250 mL örnek çözeltileri
(sırasıyla 4, 2, 1 ve 0.4 µg/L Hg(II) ve MeHg içeren) belirlenen en uygun koşullarda
kolonlardan geçirildi. En fazla 50 mL’lik örnek çözeltiden Hg(II) ve MeHg’ nin geri
kazanılabildiği görülmüştür. Daha yüksek hacim değerlerinde geri kazanma verimleri
gittikçe azalmaktadır. 50 mL örnek içerisindeki Hg(II) ve MeHg’nın 10 mL geri
alma çözeltisine tamamen alındığı kabul edilirse her iki tür için de 5 katlık bir
derişim artışı (zenginleşme) söz konusudur. Bu koşullarda yöntemin zenginleştirme
faktörü 5 olarak bulunur.
Şekil 4.4. Örnek hacminin cıva türlerinin geri kazanma verimine etkisi
Yabancı iyonların geri kazanım verimine etkisi (Girişim etkisi)
Geliştirilen yöntemin uygulanabilirliği araştırılırken en önemli değişkenlerden biri de
girişim etkisi yapan iyonların tespit edilmesidir. Bu amaçla Na+, K+, Ca2+, Mg2+,
Ni(II), Cu(II), Zn(II), Cr(III), Fe(III), Mn(II), Co(II), As(III) ve Se(IV) gibi bazı
iyonların Hg(II) ve MeHg’ nin zenginleştirilmelerine etkileri incelenmiştir. Bu
amaçla Hg(II) ve MeHg derişimleri sabit tutularak diğer iyonların derişimleri giderek
artırıldı ve geri kazanma verimin % 90’nın üstünde olduğu değerler tespit edildi.
Sonuçlar Çizelge 4.3 ve 4.4’de verilmiştir.
62
Çizelge 4.3. Hg(II)’ nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde yabancı
iyonların etkisi (Model çözelti 4 ng/ml)
İyon
+
Na
K+
Ca2+
Mg2+
Mn(II)
İyon/Analit
Derişim, mg/L
%Geri Kazanma Verimia
1250
5
101±3
2500
10
98±4
6250
25
103±4
12500
50
101±3
25000
100
97±3
62500
250
94±3
125000
500
81±3
1250
5
98±4
2500
10
101±3
6250
25
99±3
12500
50
96±2
25000
100
95±3
62500
250
86±4
1250
5
98±3
2500
10
105±2
6250
25
101±3
12500
50
95±4
25000
100
87±3
1250
5
97±2
2500
10
95±4
6250
25
103±2
12500
50
99±3
25000
100
84±3
1250
5
98±3
2500
10
96±2
6250
25
96±4
12500
50
103±4
63
Çizelge 4.3.(Devam) Hg(II)’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi (Model çözelti 4 ng/ml)
İyon
İyon/Analit
Derişim, mg/L
% Geri KazanmaVerimia
Mn(II)
25000
100
101±3
62500
250
83±2
1250
5
98±3
2500
10
95±2
6250
25
96±4
12500
50
94±3
25000
100
82±4
1250
5
101±2
2500
10
98±4
6250
25
99±2
12500
50
102±3
25000
100
108±4
62500
250
84±3
1250
5
102±3
2500
10
98±4
6250
25
105±3
12500
50
101±2
25000
100
103±5
62500
250
78±4
1250
5
98±2
2500
10
96±3
6250
25
97±4
12500
50
89±3
1250
5
97±3
2500
10
95±4
6250
25
98±3
12500
50
87±3
Ni(II)
Cu(II)
Cr(III)
Co(II)
Cd(II)
64
Çizelge 4.3.(Devam) Hg(II)’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi (Model çözelti 4 ng/ml)
İyon
Derişim, mg/L
1250
5
102±5
2500
10
97±3
6250
25
98±3
12500
50
94±4
25000
100
86±3
1250
5
97±3
2500
10
95±4
6250
25
78±3
1250
5
101±3
2500
10
102±4
6250
25
96±3
12500
50
89±3
1250
5
96±3
2500
10
97±1
6250
25
93±3
12500
50
87±3
Zn(II)
Fe(III)
As(III)
Se(IV)
a
% Geri KazanmaVerimia
İyon/Analit
Üç ölçümün ortalaması
Çizelge 4.4. MeHg’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde yabancı
iyonların etkisi (Model çözelti 4 ng/ml)
İyon
Na+
K+
İyon/Analit
Derişim, mg/L
% Geri KazanmaVerimia
1250
5
97±3
2500
10
98±4
6250
25
96±3
12500
50
91±3
25000
100
86±5
1250
5
98±5
2500
10
96±3
65
Çizelge 4.4.(Devam) MeHg’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi (Model çözelti 4 ng/ml)
İyon
K+
Ca2+
Mg2+
Mn(II)
Ni(II)
Cu(II)
Cr(II)
İyon/Analit
Derişim, mg/L
% Geri KazanmaVerimia
6250
25
97±4
12500
50
93±3
25000
100
83±4
1250
5
95±3
2500
10
101±4
6250
25
94±2
12500
50
90±3
25000
100
78±3
1250
5
97±4
2500
10
98±3
6250
25
95±3
12500
50
91±2
25000
100
80±3
1250
5
98±3
2500
10
96±5
6250
25
93±4
12500
50
81±5
1250
5
95±3
2500
10
96±4
6250
25
92±3
12500
50
78±3
1250
5
97±2
2500
10
95±3
6250
25
90±3
12500
50
83±4
1250
5
96±3
2500
10
94±4
6250
25
92±3
66
Çizelge 4.4.(Devam) MeHg’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesinde
yabancı iyonların etkisi (Model çözelti 4 ng/ml)
İyon
İyon/Analit
Derişim, mg/L
% Geri KazanmaVerimia
Cr(III)
12500
50
82±5
1250
5
98±3
2500
10
97±4
6250
25
92±3
12500
50
83±4
1250
5
99±3
2500
10
95±3
6250
25
90±5
12500
50
76±4
1250
5
97±2
2500
10
94±3
6250
25
88±4
1250
5
98±3
2500
10
95±3
6250
25
83±4
1250
5
99±2
2500
10
95±3
6250
25
90±4
1250
5
98±2
2500
10
94±4
6250
25
87±3
Co(II)
Cd(II)
Zn(II)
Fe(III)
As(III)
Se(IV)
a
Üç ölçümün ortalaması
67
4.1.2. Yöntemin analitik başarımı
Gözlenebilme Sınırı ve Tayin Sınırı
Aletsel gözlenebilme sınırını belirleyebilmek için pH’sı 4’e ayarlandı ve cıva sinyali
gözleyebilmek için içine az miktarda Hg(II) eklenmiş tanık numune belirlenen en
uygun kolonlarda kolondan geçirildi ve zenginleştirme yapılmadan 50 mL geri alma
çözeltisiyle geri alma işlemi yapıldı. Tanık numune sinyallerinin (N=20) standart
sapmasının üç katına karşılık gelen aletsel gözlenebilme sınırı (LOD) eşitlik 2.10 ile
hesaplandı. Tanık numune sinyallerinin standart sapmasının 10 katına karşılık gelen
tayin sınırı da (LOQ) eşitlik 2.11 ile hesaplanmıştır. Hesaplanan LOD ve LOQ
değerinin zenginleştirme faktörüne bölünmesi ile analitik gözlenebilme ve analitik
tayin sınırı hesaplanmıştır. Aynı işlemler MeHg için de yapılmıştır. Elde edilen
sonuçlar Çizelge 4.5’de verilmiştir.
Kesinlik
Yöntemin kesinliğinin belirlenmesi amacıyla belirlenen en uygun koşullarda Hg(II)
ve MeHg iyonlarının zenginleştirme işlemi örnek çözeltisinin kolondan geçirilmesi,
tutunan türlerin geri alınması ve yıkama 5 kez tekrarlandı ve geri kazanma verimleri
hesaplandı. Ortalama geri kazanma verimleri ve geri kazanma verimlerinin bağıl
standart sapması hesaplanarak yöntemin kesinliği bulunmuştur. Elde edilen sonuçlar
Çizelge 4.5’de verilmiştir.
Kalibrasyon grafiği ve doğrusal çalışma aralığı
Hg(II) ve MeHg iyonları için CVAAS ile elde edilen kalibrasyon grafiği Şekil 4.5 ve
Şekil 4.6’da verilmiştir. Doğrusal çalışma aralığının alt sınırı LOQ değeri olarak
alınmış, üst sınırı kalibrasyon doğrusunun doğrusallıktan saptığı derişim olarak
belirlenmiştir. Elde edilen sonuçlar Çizelge 4.5’de verilmiştir.
68
Şekil 4.5. Hg(II) için kalibrasyon grafiği
Şekil 4.6. MeHg için kalibrasyon grafiği
Doğruluk
Yöntemin doğruluğunu araştırmak amacıyla belgeli referans madde (CRM) analizi
yapılmıştır. Sonuçlar Çizelge 4.5’de verilmiştir. Çizelge 4.5’de görüldüğü gibi
belgeli değer MeHg için -% 7,8 bağıl hata ile tayin edilmiştir Hg(II) için ise değer
tayin sınırının altındadır. Bu hata analitik açıdan kabul edilebilir düzeydedir.
69
Yöntemin doğruluğu belgeli referans madde ile gösterilmesine karşılık, gerçek
örneklerdeki analizler sırasında da katkılı örnek analizleri ile doğruluk bir kez daha
kontrol edilmiştir.
Çizelge 4.5. Hg(II) ve MeHg’nin Amberlyst 36 dolgulu kolonda zenginleştirilmesi
ve türlemesi için bulunan analitik başarım ölçütleri
Analitik başarım ölçütü
Hg(II)
LOD, (N=20), (µg/L)
2,2
2,8
(LOD/Z.F) (µg/L), (Z.F=5)
0,44
0,56
LOQ, (N=20), (µg/L)
7,3
15,2
Analitik tayin sınırı (LOQ/Z.F)
1,5
3,0
%99
%99
Bağıl standart sapma %
±3
±2
Deney sayısı (N)
5
5
y = 0,0034C+0,0057
y = 0,0067C-0,009
0,9939
0,9929
0,44 - 50
0,56 – 50
0,146
5,395
µg/L
0,292
10,79
Bulunan değer (N=3)
TSA
4,97
-
-%7,8
MeHg
Analitik gözlenebilme sınırı
(µg/L), (Z.F=5)
Ortalama geri kazanma
Kesinlik
verimi
Kalibrasyon denklemi
Doğru
C=µg/L
Denklemi r2
Doğrusal çalışma aralığı
(µg/L)
ERM-CE464 Belgeli
Doğruluk
değer (mg/kg)
Çözeltideki derişimi,
Bağıl hata %
(ZF: Zenginleştirme Faktörü, TSA: Tayin Sınırının Altında, LOD: Gözlenebilme
Sınırı, LOQ: Tayin Sınırı)
70
4.1.3. Model çözeltide cıva tayini ve türlemesi
Bölüm 3.3.1 de verildiği şekilde hazırlanan model çözeltilere önerilen zenginleştirme
ve türleme yöntemi uygulanarak cıva türleri tayin edilmiştir. Bulunan sonuçlar
Çizelge 4.6 da verilmiştir. Model çözeltiye eklenen Hg(II) ve MeHg iyonları oldukça
küçük bağıl hata ile tayin edilmiştir. Örnekte tek başına Hg(II) ve MeHg
bulunduğunda -%2,5 her iki türde -%2,5 tayin edilebilmiştir. Her iki tür birlikte
bulunduğunda ise Hg(II) %5,0,
MeHg türü de -%5,0 bağıl hata ile tayin
edilebilmiştir. Toplam cıva tayinindeki hata % 10’dan küçüktür.
4.1.4. Su örneklerinde cıva tayini ve türlemesi
Bölüm 3.3.2 de verildiği şekilde hazırlanan su örneklerine önerilen zenginleştirme ve
türleme yöntemi uygulanmıştır. Bulunan sonuçlar Çizelge 4.7’de verilmiştir.
Doğruluğu da kontrol etmek için katkılı su örneklerinde de cıva türlerinin tayini
yapılmıştır. Çizelge 4.7’de görüldüğü gibi bağıl hatalar analitik açıdan kabul
edilebilir düzeydedir.
4.1.5. Balık örneğinde cıva tayini ve türlemesi
Balık örneğinde cıva tayini yapmak için kurutulan balık örneğinden 1 g alınarak
bölüm 3.3.3’de verildiği gibi örnek çözeltileri hazırlandı. Çözeltilere deneysel olarak
belirlenen en uygun koşullarda zenginleştirme ve türleme yöntemi uygulandı.
Bulunan sonuçlar Çizelge 4.8’de verilmiştir. Bağıl hatalar analitik açıdan kabul
edilebilir düzeydedir.
77 71
Çizelge 4.6. Model çözelti ile cıva türleme çalışması
Örnek
Numarası
a
Bulunana, µg/L
Eklenen, µg/L
Hg(II) MeHg
MeHg
(Hg(II) cinsinden)
Hg(II)
MeHg
TSA
1
-
-
-
TSA
2
4
-
-
3,9±0,1
3
-
4
3,72
-
4
4
4
3,72
4,2±0,2
% Bağıl Hata
MeHg
Toplam
(Hg(II) cinsinden)
Hg
-
Hg(II)
-
MeHg
-
Toplam
Hg
-
-
3,9±0,1
-2,5
3,9±0,2
3,6±0,2
3,6±0,2
-
-2,5
-2,5
3,8±0,4
3,5±0,4
7,7±0,4
+5,0
-5,0
0
-2,5
Üç ölçümün ortalaması ± standart sapma, Toplam Hg = Hg(II) + MeHg (Hg(II) cinsinden)
71
72
96
Çizelge 4.7. Su örneklerindeki cıva türleri tayini (Örnek hacmi 50 mL)
Bulunana, µg/L
Eklenen, µg/L
Örnek
Hg(II)
MeHg
MeHg
(Hg(II) cinsinden)
Hg(II)
MeHg
-
-
-
TSA
TSA
4
4
3,72
3,9±0,2
Beypazarı Kaplıca
-
-
-
Suyu
4
4
Ayaş İçmece
-
Suyu
Kızılcahamam
Çeşme suyu
Kaplıca Suyu
a
% Bağıl Hata
MeHg
(Hg(II) cinsinden)
Toplam
Hg
Hg(II)
MeHg
Toplam
Hg
-
-
-
-
-
3,9±0,2
3,6±0,2
7,5±0,2
-2,5
-2,5
-2,8
TSA
TSA
-
-
-
-
-
3,72
3,9±0,1
3,8±0,4
3,5±0,4
7,4±0,3
-2,5
-5,0
-4,1
-
-
TSA
TSA
-
-
-
-
-
4
4
3,72
3,7±0,2
4,1±0,1
3,8±0,1
7,5±0,3
-7,5
+2,5
-2,8
-
-
-
TSA
TSA
-
-
-
-
-
4
4
3,72
4,1±0,1
3,9±0,2
3,6±0,2
7,7±0,2
+2,5
-2,5
-0,3
Üç ölçümün ortalaması ± standart sapma, Toplam Hg = Hg(II) + MeHg (Hg(II) cinsinden)
72
96 73
96
Çizelge 4.8. Balık örneğindeki cıva türleri tayini
Örnek
% Bağıl Hata
MeHg
(Hg(II) cinsinden)
Toplam
TSA
-
2,1± 0,3
-
-
4,2±0,2
2
1,86
-
4
6
2
7
4
Numarası
a
Bulunana, µg/L
Eklenen, µg/L
MeHg
Hg(II)
(Hg(II) cinsinden)
Hg(II)
MeHg
1
-
-
-
TSA
2
2
-
-
3
4
-
4
-
5
MeHg
Toplam
Hg(II)
MeHg
-
-
-
-
-
2,1± 0,3
+5,0
-
+5,0
-
-
4,2±0,2
+5,0
-
+5,0
-
1,9±0,2
1,8±0,2
1,8±0,2
-
-5,0
-5,0
3,72
-
4,1±0,1
3,8±0,1
3,8±0,1
-
-2,5
-2,5
2
1,86
1,9±0,2
2,1±0,1
1,9±0,1
3,8±0,2
-5,0
+5,0
-0,2
4
3,72
3,7±0,2
3.9±0,2
3,6±0,2
7,3±0,2
-7,5
-2,5
-5,1
Hg
Hg
Üç ölçümün ortalaması ± standart sapma, Toplam Hg = Hg(II) + MeHg (Hg(II) cinsinden)
73
74
4.1.6. Adsorpsiyon izotermi ve adsorpsiyon kapasitesi
Adsorbanın Hg(II) için adsorpsiyon kapasitesi Bölüm 3.4.3’de verilen yöntemle tayin
edildi. Bu amaçla 5, 10, 25, 50, 100, 200 µg/L Hg(II) içeren 50 mL’lik örnek
çözeltilerinin pH değerleri 4’e ayarlandı ve 100 mg Amberlyst 36 içeren erlene
konuldu. Belirli süre beklenerek sistemin dengeye gelmesi sağlandı ve çözeltiden bir
kısım alınarak cıva türleri tayin edildi. Denge durumunda çözeltideki cıva derişimi
(CE) ve adsorbanın gramı başına tutunan cıva türlerinin miktarı hesaplandı. Langmiur
eşitliğinin doğrusallaştırılmış haline göre CE’ye karşı CE/QE grafiği çizildi (Şekil
4.7). Şekil 4.7’de doğrunun eğim ve kesim noktasında yararlanılarak Qo ve b
Langmuir sabitleri hesaplandı. Adsorpsiyon kapasitesine karşılık gelen Qo, 57,80
mg/g olarak, Langmuir sabiti ise 0,0324 L/mg olarak hesaplanmıştır.
5
y = 0.0173x + 0.5334
R² = 0.9822
Ce/Qe (g/L)
4
3
2
1
0
0
50
100
150
200
250
Ce (mg/L)
Şekil 4.7. Hg(II) için doğrusallaştırılmış Langmuir eşitliğinin grafiği
Adsorbanın MeHg için adsorbsiyon kapasitesi aynı şekilde hesaplandı. Bu amaçla 5,
10, 25, 50, 100, 200 µg/mL MeHg içeren 50 mL’lik örnek çözeltilerinin pH değerleri
4’e ayarlandı ve 100 mg Amberlyst 36 maddesi kullanıldı. Langmiur eşitliğinin
doğrusallaştırılmış haline göre CE’ye karşı CE/QE grafiği çizildi. Şekil 4.8’den
75
doğrunun eğim ve kesim noktasında yararlanılarak Qo ve b Langmuir sabitleri
hesaplandı. Adsorpsiyon kapasitesine karşılık gelen Qo, 58,13 mg/g olarak, Langmuir
sabiti ise 0,0462 L/mg olarak hesaplanmıştır.
Şekil 4.8. MeHg için doğrusallaştırılmış Langmuir eşitliğinin grafiği
76
5. TARTIŞMA
Eser düzeydeki elementlerin derişimlerinin tayininde, kullanılan yöntemin tayin
sınırının altında olması veya çeşitli girişim etkileri sebebiyle genellikle istenilen
duyarlılıkta doğrudan tayinleri yapılamamaktadır. Bu nedenle tayinlerde girişimden
kaynaklanan problemlerin giderilmesi veya tayin sınırını düşürülmesi için bir
zenginleştirme / ayırma yöntemine ihtiyaç duyulmaktadır. Zenginleştirme işlemi ile
tayin edilecek eser element, büyük hacimli bir örnekten, bağıl olarak oldukça küçük
hacimli bir tayin çözeltisi içine alınır. Bu sırada girişim oluşturabilecek bileşenler
ortamdan uzaklaştırılarak ayırma işlemi de gerçekleştirilir. Sonuçta tayin edilecek
eser element, bileşimi bilenen ve örneğe göre daha basit bir ortama alınmış olur. Bu
işlemlerle tayin edilecek eser element, girişim yapan türlerden ayrılmış ve örneğe
göre daha basit bir ortama alınmış olur.
Bu tez kapsamında, Amberlyst 36 eser düzeydeki Hg(II) ve MeHg’nin kolon
tekniğiyle
zenginleştirilmesi
ve
türlemesinde
katı
faz
özütleyicisi
olarak
kullanılmıştır. Bu çalışmada Hg(II) ve MeHg karmaşık örnek ortamlarından
ayrılması, türlemesi ve zenginleştirilmesi için uygun koşullar belirlenmiştir. Bazı
elementlerin toplam derişiminden çok o ortamda elementin hangi türü olarak
bulunduğu da önemlidir. Çünkü türlerden biri toksik olmadığı halde diğeri
olabilmektedir. Bu kapsamda geliştirilen yöntem Hg(II) ve MeHg iyonlarının
türlemesi için uygulanmıştır. Önerilen yöntem ile Hg(II) ve MeHg iyonları aynı
pH’da farklı geri alma çözeltileri kullanılarak ayrılmıştır. Deneysel olarak pH 4 ‘te
MeHg iyonları 10 mL 0,1 mol/L HCl kullanılark, Hg(II) iyonları ise 3 mol/L HCl
içinde 0,2 mol/L tiyoüre çözeltisi kullanılarak geri alınmıştır. Toplam cıva miktarı bu
iki türün Hg(II) cinsinden miktarlarının toplanmasıyla bulunmuştur.
Eser elementlerin zenginleştirilmesi ve türlemesinde geri kazanma verimini etkileyen
en önemli değişken ortamın pH’sıdır. Ortamın pH değerindeki değişmeye bağlı
olarak adsorbanın yapısı değişmekte bağlayıcı grupların sayısı artmakta veya
azalmaktadır. Geliştirilen yönteme göre civanın türleme ve zenginleştirilmesinde pH
4’te her iki tür için de en yüksek geri kazanım değerinin elde edildiği pH olmuştur.
77
Cıva türlerinin çalışma aralığının asidik bölgede olması örnek ortamının kararlılığı
açısından olumludur. Çünkü bazik ortamlarda örnek ortamında bulunabilecek metal
iyonları hidroksitleri halinde çökerek ortamın fiziksel yapısını etkileyebilirler. Bazik
bölgede elementlerin hidroksitler halinde çökerek verdikleri çökelek üzerine tayin
elementi tutunabilir bu durumda katı faz olarak kullanılan Amberlyst 36 üzerinde
tutunmadan söz edilemez. Gerçek örneklerin çözülmesi ve iyileştirilmesi genelde
asidik ortamda yapıldığından elde edilen son çözelti asidik olmakta ve pH ayarlaması
daha kolay yapılabilmektedir.
Adsorpsiyon ile zenginleştirmede geri kazanma verimine etki eden iki olaydan biri,
zenginleştirilecek elementin kolonda tutunması, diğeri de kolonda tutunan elementin
geri alınmasıdır. Bu iki işlemden herhangi biri yeteri kadar yapılamazsa, geri
kazanma verimi istenen değere ulaşmaz. Bu nedenle yapılan çalışmada bir taraftan
tutunma şartları en uygun hale getirilirken diğer taraftan da geri alma koşulları en
uygun hale getirilmelidir. Zenginleştirme konusunda daha önce yapılan çalışmalar
incelendiğinde geri alma çözeltisi olarak genellikle asitlerin, bazların veya bazı
organik çözücülerin kullanıldığı görülmüştür. Bu çalışmada kolonda tutunan
elementleri geri almak için öncelikle yaygın kullanılan inorganik asitler denenmiştir.
Denenen asit çözeltileriyle MeHg için yeterli yeterli geri kazanma verimine (> %95)
ulaşılmış ancak Hg(II) için ise asit çözeltisinde organik tuz (tiyoüre) çözülerek geri
alma çözeltisi olarak kullanılmış ve yeterli geri kazanma verimi (>%95) elde
edilmiştir. Son örnek çözeltisinin asidik olması CVAAS yönteminde tayinlerde
uygun ortamlardır. Tayin aşamasında ayrıca örneğin asitlendirilmesine gerek
kalmamaktadır.
Zenginleştirmede önemli deneysel değişkenlerden biri de örneğin kolondan geçme
süresidir. Geçiş süresi bize temas süresinin tayin edilecek türün geri kazanma
verimine etkisi bakımından bilgi vermektedir. Hızlı geçişlerde adsorbanla temas
süresi azalacağından geri kazanma veriminde de azalmalar beklenir. Bu çalışmada
MeHg ve Hg(II) iyonları 3 mL/min aynı akış hızında kolondan geçirilerek geri
alınabilmiştir. Akış hızının aynı olması aynı örnek çözeltinin kolonda geçirilip
türlerin birbirinden ayrılması açısından önemlidir.
78
Yüksek zenginleştirme faktörü elde edebilmek için geri alma çözeltisinin hacminin
küçük olması gerekir. Bazı durumlarda küçük geri alma çözelti hacmi, adsorbanda
tutunan elementlerin geri kazanılmasında yeterli olmayabilir. Bu gibi durumlarda
adsorban yüksek asit ya da baz derişiminden etkilenmiyorsa, geri alma çözeltisinin
derişimi artırılabilir. Yukarıda bahsedilen tüm ölçütler dikkate alınarak, en uygun
geri alma çözeltisi MeHg için 10 mL 0,1 mol/L HCl çözeltisi, Hg(II) için ise 10 mL
3 mol/L HCl içerisinde 0,2 mol/L tiyoüre çözeltisi kullanılmıştır. Zenginleştirilecek
elementin örnek çözeltisindeki derişimi çok düşük olduğunda, tayinin yapılabilmesi
için oldukça büyük hacimdeki örnek çözeltisinin kolondan geçirilmesi gerekir. Çok
seyreltik çözeltilerin büyük hacimlerinin kolondan geçirilmesiyle zenginleştirmenin
yapılıp yapılmayacağını araştırmak için örnek hacminin geri kazanma verimine etkisi
de incelenmiştir. Bu amaçla model çözeltideki element miktarı sabit tutularak çözelti
hacmi artırılmış ve böylece gittikçe azalan derişimlerde çözeltiler hazırlanmıştır. Bu
çalışma, örnek hacmi yanında derişimin etkisini de göstermektedir. Aynı miktarda
element içeren farklı hacimdeki çözeltilerde derişim de farklı olmaktadır. Belirlenen
en uygun şartlarda yapılan zenginleştirme işlemi sonunda MeHg ve Hg(II) için 50
mL’ ye kadar zenginleştirilmiştir. Elde edilen bu sonuçlara göre, 50’er mL MeHg ve
Hg(II) çözeltisi kolondan süzüldüğünde ve bu elementler kolondan 10 mL’lik geri
alma çözeltileri ile geri kazanıldığında zenginleştirme katsayısı kuramsal olarak,
5’dir. Bu çalışmada zenginleştirme faktörü oldukça düşüktür, fakat cıva türlerinin
karmaşık ortamdan ayrılması ve türlendirilmesinin yapılabilmesi önemlidir.
Geliştirilen yöntemin kesinliğini araştırmak için model çözeltilerle zenginleştirme
işlemi 5 kez tekrarlanmış ve elde edilen sonuçlara göre ortalama yüzde geri kazanma
verimleri ve bağıl standart sapma değerleri belirlenmiştir. Hg(II) için % 99 geri
kazanma verimi ve ±3 bağıl standart sapma MeHg için ise % 99 geri kazanma verimi
±2 bağıl standart sapma değerleri bulunmuştur. Ancak bu çalışma model çözelti
ortamında yapılmıştır. Gerçek örnek ortamında bağıl standart sapmanın daha yüksek
olması beklenebilir. Bu sonuç gerçek örnek analizlerinde görülmektedir (Çizelge 4.7
ve 4.8)
79
Doğruluk kontrolü için Belgeli Referans Maddesi kullanılmış ve içeriği MeHg için %7,8 bağıl hata ile tayin edilmiştir. Hg(II) için ise değer tayin sınırının altında
olduğundan tayin edilememiştir. Gerçek örnek ortamına sahip bu belgeli referans
maddelerde yapılan zenginleştirme, yöntemin gerçek örneklerde girişim etkisi
olmadan kullanılabileceğini göstermektedir.
Genellikle katı faz olarak kullanılan adsorbanların tayin elementini ortamda bulunan
diğer elementlerden ayırarak onların girişim etkisini azaltması veya yok etmesi
beklenir. Adsorbanlar bir elementi seçimli olarak tutuyorsa, o elementi ana
bileşenlerden ayırarak onların girişim etkisini yok eder. Ortamda bulunan her bir
element adsorbana farklı şartlarda (pH, akış hızı vb.) en yüksek verimle tutunur. Bu
elementlerin kolondan geri alınma şartları da farklı olduğu için, en uygun şartlar
sağlanarak seçimlilik sağlanabilir. Bu çalışmada yabancı iyonların Hg(II)’nın geri
kazanma verimine etkisi Na+ için 250 mg/L; K+, Mn(II), Cu(II) ve Cr(III) için 100
mg/L; Ca2+, Mg2+, Ni(II), Zn(II) için 50 mg/L; Co(II), Cd(II), As(III) , Se(IV) için 25
mg/L; Fe3+ için ise 10 mg/L derişime kadar girişim önemli değildir. Yabancı
iyonların MeHg’ nin geri kazanma verimine etkisi ise, Na+, K+, Ca2+ ve Mg2+ için 50
mg/L; Mn(II), Ni(II), Cu(II), Cr(III), Co(II) ve Cd(II) için 25 mg/L; Zn(II), Fe(III),
As(III) ve Se(IV) için ise 10 mg/L derişime kadar girişim önemli değildir. Bu oranlar
bize yöntemin çoğu çevre örneğinde bile girişime maruz kalmadan kullanılacağını
göstermektedir.
Yöntemin gerçek örneklere uygulanabilirliğini incelemek amacı ile Ankara
çevresinden temin edilen çeşitli kaplıca suyu örneklerinde ve balıkta zenginleştirme
ve türleme çalışmaları yapılmıştır. Çizelge 4.7 ve Çizelge 4.8’de verilen sonuçlara
göre cıva türleri % 10’dan daha düşük bağıl hata ile tayin edilebilmektedir.
Geliştirilen yöntemin, aletsel gözlenebilme sınırı (LOD), MeHg için 2,8 µg/L Hg(II)
için ise 2,2 µg/L olarrak bulunmuştur. Bu değerlerin zenginleştirme faktörlerine
bölünmesi ile yöntemin analitik gözlenebilme sınırı bulunmaktadır. Analitik
gözlenebilme sınırları ise metil cıva için 0,44 µg/L, inorganik cıva için ise 0,56 µg/L
olarak bulunmuştur.
80
Kolon dolgu maddesinin tayin edilecek elementi adsoplama yeteneği adsorplama
kapasitesi ile verilmektedir. Doğrusallaştırılmış Langmuir izoterminin eğiminden
bulunan bu değer adsorbanın gramı başına tutunan miktarı (mg/g) olarak
verilmektedir. Kullanılan adsorbanın cıva türleri için adsorpsiyon kapasitesi, kesikli
yöntemle çalkalamalı su banyosunda yapılmıştır. Kesikli yöntemle, oda şartlarında
kullanılan Amberlyst 36 malzemesinin MeHg ve Hg(II) için adsorpsiyon kapasitesi
sırasıyla 57,80 ve 58,13 mg/g olarak bulunmuştur.
Cıva tayini için geliştirilen yöntemin literatürde verilen bazı zenginleştirme ve
türleme çalışmaları ile karşılaştırması Çizelge 4.9’da verilmiştir.
81
Çizelge 4.9. Cıvanın Amberlyst 36 ile zenginleştirilmesi ve türlemesi çalışmasının literatürdeki diğer çalışmalarla
karşılaştırılması
Örnek
Hg Türü
Zenginleştirme tekniği
LOD, µg/L
Referans
CVAAS
0,012
CVAAS
0,02
[Pourreza ve ark.,
2009]
[Pourreza ve
Ghanemi,2009]
ICP-AES
0,09
[Zhang ve ark.,
2010]
CVAAS
0,05
[Liu ve ark.,2005]
CVAAS
0,014
CVAAS
MeHg, 0,56
Hg(II), 0,44
[Haji Shabani ve
ark., 2004]
Bu çalışma
Tayin
yöntemi
Su, deniz ürünleri
Su, balık
Su, balık
Su, sediment
Su
Su, balık
Hg(II)
N-(klorobenzil)-N’
feniltiyoüre-sülfür tozu, SPE
Hg(II)
2-merkaptobenzimidazol ile
modifiye edilmiş agar, SPE
Hg(II)
2,6 piridin dikarboksilik asit
ile immobilize edilmiş nano
silika, (SiO2-2,6-PDCA),
SPE
Hg(II)
Hg(II) baskılıdiazoaminobenzenvinilpiridin kopolimer, SPE
Hg(II)
Ditizon immobilize
mikrokristalin naftolen, SPE
MeHg, Hg(II) Amberlyst 36, SPE
81
82
KAYNAKLAR
Alfassi, Z.B., Wai, C.M., “Preconcentration techniques for trace elements”, CRC
Press, Boca Raton, (1992).
Alonso, E.V., Siles Cordero, M.T., Torres, A.G., Rudner, P.C., Cano Pavón, J.M.,
“Mercury speciation in sea food by flow injection cold vapor atomic absorption
spectrometry using selective solid phase extraction”, Talanta, 77: 53–59 (2008).
Berkkan, A., “Tuz içeren farmasötik preparatlarda akışa enjeksiyonlu hidrür
oluşturmalı atomik absorpsiyon spektrometri (FI-HG-AAS) ile kurşun tayini”,
Doktora tezi, Gazi Üniversitesi Eczacılık Fakültesi, Ankara, (2004).
Chen, D., Huanga, C., Hea, M., Hua, B., “Separation and preconcentration of
inorganic arsenic species in natural water samples with 3-(2-aminoethylamino)
propyltrimethoxysilane modified orderedmesoporous silica micro-column and their
determination by inductively coupledplasma optical emission spectrometry”, J.
Hazard. Mater., 164: 1146–1151 (2009).
Chen, J., Teo, K.C., “Determination of cadmium, copper, lead and zinc in water
samples by flame atomic absorption spectrometry after cloud point extraction”, Anal.
Chim. Acta, 450: 215–222 (2001).
Çekiç, S.D., Filik, H., Apak, R., “ Use of an o –aminobenzoic acid-functionalized
XAD-4 copolymer resin for the seperation and preconcentration of heavy metal (II)
ions”, Anal. Chim. Acta, 505(1): 15-24 (2004).
Çıtak, D., “Katı faz ekstraksiyonu, birlikte çöktürme ve bulutlanma noktası
ekstraksiyonu ile bazı metallerin zenginleştirilmesi ve türlemesi”, Doktora Tezi,
Gaziosmanpaşa Üniversitesi Fen Bilimleri Enstitüsü, Tokat, 25-33 (2010).
Dos Santos, W.N.L., Dias, F.D.S., Fernandes, M.S., Rebouças, M.V., Vale, M.G.R.,
Ferreria, S.L.C., “Application of multivariate technique in method development for
direct determination of copper in petroleum condensate using graphite furnace
otomic absorption spectrometry”, J. Anal. Atom. Spectrom., 20: 127-129 (2005).
Dressler, V.L., Santos, C.M.M., Antes, F.G., Bentlin, F.R.S., Pozebon, D., Pozebon,
E.M.M., “Total Mercury, Inorganic Mercury and Methyl Mercury Determination in
Red Wine”, Food Anal. Method., 5: 505–511 (2012).
Ebdon, L., “An introduction to atomic absorption spectroscopy”, Heyden, London,
25 (1982).
Erdoğan, H., “Arsenik ve selenyumun katı faz özütleme yöntemiyle
zenginleştirilmesi, türlemesi ve atomik absorpsiyon spektroskopisi ile tayini”,
Yüksek Lisans Tezi, Gazi Üniversitesi Fen Bilimleri Enstitüsü, Ankara, 1-47
(2012).
83
Gao, Y., Shi, Z., Long, Z., Wu, P., Zheng, C, Hou, X ., “Determination and
speciation of mercury in environmental and biological samples by analytical atomic
spectrometry”, Microchem. J., 103: 1–14 (2012).
Grindlay, G., Mora, J., Gras, L., De Loos-Vollebregt, M.T.C., “Atomic spectrometry
methods for wine analysis: A critical evaluation anddiscussion of recent
applications”, Anal. Chim. Acta, 691: 18–32 (2011).
Haji Shabani, A.M., Dadfarnia, S., Nasirizadeh, N., “Speciation analysis of mercury
in water samples by cold vapor atomic absorption spectrometry after
preconcentration with dithizone immobilized on microcrystalline naphthalene”, Anal
Bioanal Chem, 378 : 1388–1391 (2004).
Haswell, S.J., “Atomic absorption spectrometry”, Elsevier Science Publishers B. V.,
Netherlands, 310 (1991).
Janoš, P., Štulík, K., Pacáková, V., “An ion-exchange separation of metal cations on
a C-18 column coated with dodecylsulphate”, Talanta, 39(1): 29-34 (1992).
Kalfa, O.M., “Nano boyutta adsorban madde ile eser elementlerin
zenginleştirilmesi”, Doktora tezi, Gazi Üniversitesi Fen Bilimleri Enstitüsü,
Ankara, 1-3, (2009).
Kalfa, O.M., Yalçınkay, Ö., Turker, A.R., “Synthesis of nano B2O3/TiO2 composite
material as a new solid phase extractor and its application to preconcentration and
separation of cadmium”, J. Hazard. Mater., 166: 455–461, (2009).
Kendüzler, E., “Bazı eser elementlerin Ambersorb 572 ile zenginleştirme şartlarının
araştırılması ve alevli atomik absorpsiyon spektrometrik yöntemle tayini”, Doktora
Tezi, Gazi Üniversitesi Fen Bilimleri Enstitüsü, Ankara, 5-20 (2003).
Kendüzler, E., Türker, A.R., “Atomic absorption spectrophometric determination of
trace copper in waters, aluminium foil and tea samples after preconcentration with 1nitroso-2-naphthol-3,6-disulfonic acid on Ambersorb 572”, Anal. Chim. Acta, 480:
259-266 (2003).
Krishna, M.V.B, Karunasagar, D., Rao, S.V., Arunachalam, J., “Preconcentration
and speciation of inirganic and methyl mercury in waters using polyaniline and gold
trap- CVAAS.” Talanta, 68: 329-335 (2005).
Leemakers, M., Baeyens, W., Quevauviller, F., Horvat, M., “Mercury in
environmental samples : Speciation, artifacts and validation”, Trend. Anal. Chem.,
24(5): 383-393 (2005).
Lemos, V.A., Teixeira, L.S.G., Bezerra, M.A., Costa, A.C.S., Castro, J.T., Cardoso,
L.A.M., Jesus, D.S., Santos, E.S., Baliza, P.X., Santos, L.N., “New Materials for
84
solid-phase extraction of trace elements”, Applied Spectroscopy Reviews, 43: 303334 (2008).
Leopold, K., Foulkes, M., Worsfold, P., “Methods for the determination and
speciation of mercury in natural waters-A review”, Anal. Chim. Acta, 663: 127–138
(2010).
Liu, Y., Chang, X., Yang, D., Guo, Y., Meng, S., “Highly selective determination of
inorganic
mercury(II)
after
preconcentration
with
Hg(II)-imprinted
diazoaminobenzene–vinylpyridine copolymers”, Anal. Chim. Acta, 538: 85–91
(2005).
Mizuike, A., “Enrichment techniques for inorganic trace analysis”, Springer-Verlag,
BerlinHeidelberg, Newyork, 5-6 (1983).
Mladenova, E.K., Dakova I.G., Tsalev, D.L., Karadjova, I.B., “Mercury
determination and speciation analysis in surface waters” Cent. Eur. J. Chem, 10(4):
1175-1182 (2012).
Mousavi, H.Z., Asghari, A., Shirkhanloo, H., “Determination of Hg in Water and
Wastewater Samples by CV-AAS Following Online Preconcentration with Silver
Trap”, J. Anal. Chem., 65: 935–939 (2010).
Oliveira, C.C., Sartini, R.P., Zagatto, E.A.G., “Microwave-assisted sample
preparation in sequential injection: spectrophotometric determination of magnesium,
calcium and iron food”, Anal. Chim. Acta, 413: 41-48 (2000).
Poole, C. F., Poole, S. K., “Chromatography today”, Elseiver, Amsterdam (1997).
Pourreza, N., Parham, H., Kiasat, A.R., Ghanemi, K., Abdollahi, N., “Solid phase
extraction of mercury on sulfur loaded with N-(2-chlorobenzoyl)-N_-phenylthiourea
as a new adsorbent and determination by cold vapor atomic absorption
spectrometry”, Talanta, 78: 1293–1297 (2009).
Pourreza, N., Ghanemi, K., “Determination of mercury in water and fish samples by
cold vapor atomic absorption spectrometry after solid phase extraction on agar
modified with 2-mercaptobenzimidazole”, J. Hazard. Mater., 161: 982–987 (2009).
Pozebon, D., Dressler, V.L., Gomes Neto, J.A., Curtius, A.J., “Determination of
arsenic(III) and arsenic(V) by electrothermal atomic absorption spectrometry after
complexation and sorption on a C-18 bonded silica column”, Talanta, 45(6): 11671175 (1998).
Rı´o-Segade, S., Bendicho, C., “Ultrasound-assisted extraction for mercury
speciation by the flow injection–cold vapor technique”, J. Anal. At. Spectrom., 14:
263–268 (1999).
85
Robinson, J.W., “Atomic Spectroscopy”, Marcel Dekker, Inc, New York, (1990).
Sarıca, Y.D., “Bazı organic bileşiklerin tepe boşluklu tek damla mikro özütleme ve
grafit fırınlı atomik absorpsiyon spektrometri ile seçimli tayini için yöntem
geliştirilmesi”, Doktora Tezi, Gazi Üniveristesi, Fen Bilimleri Enstitüsü,
Ankara,49-84 (2011).
Sarıca, Y.D., Türker, A.R., “Speciation and Determination of Inorganic Mercury and
Methylmercury by Headspace Single Drop Microextraction and Electrothermal
Atomic Absorption Spectrometry in Water and Fish”, Clean – Soil, Air, Water,
40(5): 523–530 (2012).
Sharma, R.K., Pant, P., “Preconcentration and determination of trace metal ions from
aqueous samples by newly developed gallic acid modified Amberlite XAD-16
chelating resin”, J. Hazard. Mater., 163: 295-301 (2009).
Skoog, D.A., West, D.M., Holler, F.J., “Analitik Kimya Temelleri, Cilt 2”, Bilim
Yayıncılık, Ankara, 214, 611-615, (1999).
Susan, C., Hight, J.C., “Determination of total mercury in seafood by cold vaporatomic absorption spectroscopy (CVAAS) after microwave decomposition”, Food
Chem., 91: 557–570 (2005).
Soylak, M., Ünsal, Y.E., “Chromium and iron determinations in food and herbal
plant samples by atomic absorption spectrometry after solid phase extraction on
single-walled carbon nanotubes (SWCNTs) disk”, Food Chem. Toxicol, 48: 1511–
1515, (2010).
Soylak, M., Ünsal, Y.E., “Solid-phase extraction of heavy metal ions on bucky tubes
disc in natural water and herbal plant samples”, Environ Monit Assess, 181: 577–
586 (2011).
Torres, D.P., Frescura, Vera, L.A., Curtius, A.J., “Simple mercury fractionation in
biological samples by CV AAS following microwave-assisted acid digestion or
TMAH pre-treatment “, Microchem. J., 93: 206–210 (2009).
Tunçeli, A., Türker, A.R., “Flame atomic absorption spectrometric determination of
silver after preconcentration on Amberlite XAD-16 resin from thiocyanate solution”,
Talanta, 51(5): 889-894 (2000).
Tunçeli, A., Türker, A.R., “Speciation of Cr(III) and Cr(VI) in water after
preconcentration of its 1,5-diphenylcarbazone complex on amberlite XAD-16 resin
and determination by FAAS”, Talanta, 57: 1199-1204 (2002).
Türker, A.R., “New sorbents for solid-phase extraction for metal enrichment”, Clean
– Soil Air Water, 35: 548–557 (2007).
86
Türker, A.R., “Separation, preconcentration and speciation of metal ions by solid
phase extraction”, Sep. Purif. Rev., 41(3): 169-206 (2012).
Tüzen, M., Saygı, K.O., Soylak, M., “Novel solid phase extraction procedure for
gold(III) on Dowex M 4195 prior to its flame atomic absorption spectrometric
determination”, J. Hazard. Mater., 156: 591–595, (2008).
Tüzen, M., Uluozlu, Ö.D., Karaman, İ., Soylak, M., “Mercury(II) and methyl
mercury speciation on Streptococcus pyogenes loaded Dowex Optipore SD-2”, J.
Hazard. Mater., 169: 345–350 (2009).
Ünsal, Y.E., Soylak, M., “Double-walled Carbon Nanotubes as a Solid Phase
Extractor for Speciation-Preconcentration of Traces of Gold from Geological and
Water Samples”, Int. J. Environ. An. Ch., 91: 440-447 (2011).
Welz, B., Sperling, M. “Atomic Absorption Spectrometry”, Wiley-VCH Verlag
GmbH&Co. KgaA, Weinheim, (1999).
Welz, B., Becker-Ross, H., Florek, S., Heitmann, U., “High-Resolution Continuumsource AAS the beter way to do atomic absorpsiyon spectrometry”, Wiley-VCH
Verlag GmbH&Co. KgaA, Weinheim, (2005).
Welz, B., Becker-Ross, H., Florek S., Heitmann, U., Goreti, M., Vale, R., “HighResolution Continuum-source Atomic Absorption Spectrometry–What Can We
Expect”, J. Braz. Chem. Soc., 14(2): 220-229 (2003).
Yalçınkaya, Ö., “Bazı eser elementlerin Aluminyum Oksit/ Tek Duvarlı Karbon
Nanotüp ve Zirkonyum Oksit/Bor Oksit nano malzemeleri kullanılarak katı faz
özütleme tekniği ile zenginleştirilmesi ve tayini”, Doktora Tezi, Gazi Üniveristesi,
Fen Bilimleri Enstitüsü, Ankara, 5-40 (2010).
Yalçınkaya, Ö., Kalfa, O.M., Turker, A.R., “Chelating agent free-solid phase
extraction (CAF-SPE) of Co(II), Cu(II) and Cd(II) by new nano hybrid material
(ZrO2/B2O3)”, J. Hazard. Mater., 195: 332– 339 (2011).
Yıldız A., Genç, Ö., “Enstrumental Analiz”, Hacettepe Üniversitesi Yayınları,
Ankara, 191-193 (1993).
Zhang, L., Huang, T., Zhang, M., Guo, X., Yuan, Z., “Studies on the capability and
behavior of adsorption of thallium on nano-Al2O3”, J. Hazard. Mater., 157: 352–357
(2008).
Zhang, Y., Adeloju, S.B., “Speciation of mercury in fish samples by flow injection
catalytic cold vapour atomic absorption spectrometry”, Anal. Chim. Acta, 721: 22–
27 (2012).
87
Zhang, L., Chang, X., Hu, Z., Zhang, L., Shi, J., Gao, R., “Selective solid phase
extraction and preconcentration of mercury(II) from environmental and biological
samples using nanometer silica functionalized by 2,6-pyridine dicarboxylic acid”,
Microchim. Acta, 168: 79–85 (2010).
88
ÖZGEÇMİŞ
Kişisel Bilgiler
Soyadı, adı
: ÇABUK, Dilek
Uyruğu
: T.C.
Doğum yeri ve tarihi
: Altındağ, 02.08.1989
Medeni hali
: Bekar
e-posta
: dilekcabuk@gazi.edu.tr
Eğitim
Derece
Eğitim Birimi
Lisans
Erciyes Üniversitesi Fen Edebiyat
Lise
Yabancı Dil
İngilizce
Mezuniyet tarihi
Fakültesi Kimya Bölümü
2010
Etimesgut Lisesi
2006